Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Vincenzo Vestuto | -- | 2383 | 2022-12-27 09:36:05 | | | |
| 2 | Conner Chen | + 3 word(s) | 2386 | 2022-12-27 09:41:16 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Vestuto, V.; Sarno, V.D.; Musella, S.; Dona, G.D.; Moltedo, O.; Gomez-Monterrey, I.M.; Bertamino, A.; Ostacolo, C.; Campiglia, P.; Ciaglia, T. Endoplasmic Reticulum Stress in Diseases Pathogenesis. Encyclopedia. Available online: https://encyclopedia.pub/entry/39436 (accessed on 27 July 2026).
Vestuto V, Sarno VD, Musella S, Dona GD, Moltedo O, Gomez-Monterrey IM, et al. Endoplasmic Reticulum Stress in Diseases Pathogenesis. Encyclopedia. Available at: https://encyclopedia.pub/entry/39436. Accessed July 27, 2026.
Vestuto, Vincenzo, Veronica Di Sarno, Simona Musella, Giorgio Di Dona, Ornella Moltedo, Isabel Maria Gomez-Monterrey, Alessia Bertamino, Carmine Ostacolo, Pietro Campiglia, Tania Ciaglia. "Endoplasmic Reticulum Stress in Diseases Pathogenesis" Encyclopedia, https://encyclopedia.pub/entry/39436 (accessed July 27, 2026).
Vestuto, V., Sarno, V.D., Musella, S., Dona, G.D., Moltedo, O., Gomez-Monterrey, I.M., Bertamino, A., Ostacolo, C., Campiglia, P., & Ciaglia, T. (2022, December 27). Endoplasmic Reticulum Stress in Diseases Pathogenesis. In Encyclopedia. https://encyclopedia.pub/entry/39436
Vestuto, Vincenzo, et al. "Endoplasmic Reticulum Stress in Diseases Pathogenesis." Encyclopedia. Web. 27 December, 2022.
Copy Citation
The endoplasmic reticulum (ER) is a dynamic structure, playing multiple roles including calcium storage, protein synthesis and lipid metabolism. During cellular stress, variations in ER homeostasis and its functioning occur. This condition is referred as ER stress and generates a cascade of signaling events termed unfolded protein response (UPR), activated as adaptative response to mitigate the ER stress condition. UPR is activated in several disorders, including inflammation, diabetes, metabolic and neurodegenerative diseases, as well as in cancer.
ER stress
UPR
TRP channels
cancer
inflammation
1. Introduction
Transient receptor potential (TRP) channels represent key sensors for homeostasis maintenance in cellular environments, considering their capability to finely regulate ion balance and to modulate effectors activity. Dysregulation in TRP activity or expression is involved in the genesis of several pathological conditions ranging from neuropathic pain [1], neurodegenerative diseases [2], overactive bladder syndrome [3] to cancer [4]. Several efforts in the research field were made to understand the correlation between TRP channels aberrant activity and disease onset, while poor knowledge of their implication in endoplasmic reticulum (ER) stress development are reported.
2. ER Stress and UPR Overview
The ER represents one of the most functional and extended cell organelles responsible for multiple biological functions. Calcium storage, biomolecules synthesis, protein folding, post-translation modifications and transport, are some of the roles played by ER, contributing to cellular homeostasis. Moreover, ER provides critical features in intra- and intercellular signaling events, through its tubular network, harboring numerous membrane contact sites with plasma membrane, mitochondria, endosomes, Golgi apparatus, peroxisomes and lipid droplets [5][6].
Although all biological processes are finely regulated, there is a wide number of factors, such as nutrient deprivation, DNA damage, calcium depletion, oxidative stress, hypoxia, pH variations, etc., which can cause perturbance in the ER environment, resulting as the so-called ER stress that is characterized by misfolded and/or unfolded proteins accrual. The ER stress often represents a severe issue, since continuous and unresolved ER stress events can lead to cell death in response to the production of pro-apoptotic factors [7]. Nonetheless, when this state is well controlled by the cellular compensatory machinery and the perturbations are overcome, cells’ condition improves, enabling survival. The adaptive signaling response to ER stress is known as unfolded protein response (UPR) and is a self-protective mechanism acting through different routes: (1) decrease in protein translation, (2) transcription of genes encoding factors involved in ER protein folding and clearance (e.g., HRD1, BiP, SEL1L, Herp) and (3) the ER-associated protein degradation (ERAD) process by which misfolded and unfolded proteins are brought back to the cytosol, where they will subjected to the ubiquitin-proteasome system [8][9].
UPR can be decisive to determine cell fate: such as a two-faced Janus, on one side it rescues the cell from a mild or short-term stress, by restoring the ER proteostasis, whilst on the other side, if ER stress is prolonged and overwhelms the UPR, the pro-survival activity turns into a pro-apoptotic response [10][11]. More insights are required for the full understanding of the mechanisms regulating the UPR pathway. However, several studies have clearly described the link between ER stress and multiple physio-pathological states, and, particularly, the involvement of the UPR in cell stress signals and degradation pathways such as autophagy and in the ubiquitination-proteasome system [12][13].
UPR regulators consist in a set of three ER membrane-associated proteins, acting as ER stress sensors: protein kinase RNA (PKR)-like endoplasmic reticulum kinase (PERK), inositol-requiring transmembrane kinase/endoribonuclease 1 (IRE1) and activating transcription factor 6 (ATF6) (Figure 1).
These proteins contain domains located into the ER lumen, associated with the cytosolic effector domains and acting as ER stress sensors. Each UPR sensor is inactive due to the binding to the ER luminal chaperone glucose-regulated protein 78 (GRP78), also called binding immunoglobulin protein (BiP). When BiP is removed from the three ER stress transducers, it binds to misfolded and/or unfolded proteins accumulated in the ER to facilitate their folding and assembly, and to repair misfolding and aggregation. At the same time, the separation of BiP from the sensors leads both to proteins and UPR activation [14][15][16].
PERK is activated by autophosphorylation and dimerization upon ER stress. It enables a short-term response with the purpose of a pro-survival outcome by inhibiting protein translation, through phosphorylation of the α subunit of the eukaryotic translation initiation factor (eIF2α). The resulting P-eIF2α, in turn tentatively arrests most protein synthesis due to its association with the guanine nucleotide exchange factor eIF2B, by blocking its activity and, thus, causing an arrest in transient protein translation. On the other hand, the synthesis of the activating transcription factor 4 (ATF4) is increased. ATF4 is crucial for pro-survival genes activation and is aimed to maintain redox balance, given that PERK-dependent phosphorylation triggers dissociation of Nrf2/Keap1 complex. The nuclear factor erythroid 2-related factor 2 (Nrf2) represents a potent transcription factor involved in the antioxidant response. Under basal conditions, it is inactivated by the Kelch-like ECH associated protein 1 (Keap1) by degradation through the cullin3/ring box 1-depedent ubiquitin ligase complex. During ER stress, P-PERK phosphorylates Keap1 inducing conformational changes that prevent the binding of de novo produced Nrf2. Therefore, newly translated Nrf2 can migrate into the nucleus to activate antioxidant gene transcription promoting survival [17][18][19]. In the long-term, if ER stress persists and is not resolved, pro-apoptotic factors, such as C/EBP homologous protein (CHOP), are activated by ATF4, inducing the growth arrest and DNA damage-inducible protein 34 kDa (GADD34). This protein forms a complex with protein phosphatase 1 (PP1) leading to the dephosphorylation of P-eIF2α, and thus a suicidal response via the cellular protein synthesis reactivation [20][21][22] (Figure 1). Furthermore, p38 MAPK mediates phosphorylation of two adjacent serine residues (78 and 81) of CHOP, enhances its ability to function as a transcriptional activator, inducing apoptosis or different inhibitory effects [23] (Figure 2).
IRE1 is also activated by autophosphorylation and oligomerization and induces pro-survival genes to enhance ERAD and protein folding. This mechanism is allowed through IRE1 endoribonuclease activity that converts uXBP1 (unspliced X-box binding protein 1) mRNA to the transcription factor sXBP1 (spliced form) [24]. Upon prolonged ER stress, the continuous IRE1-induced RNase activity becomes less specific, resulting in a process of multiple mRNAs degradation called RIDD (regulated-IRE1 dependent decay). If ER stress is not rescued, P-IRE1 generates a complex with TNF receptor-associated factor 2 (TRAF2) and apoptotic-signaling kinase-1 (ASK1) to phosphorylate JNK (c-Jun N-terminal kinase)—in this way, apoptosis is triggered [25][26][27] (Figure 1). IRE1 signaling may also promote cell death by activating CHOP (sXBP1 pathway) (Figure 2) or caspase-12 (Figure 1), an ER-resident and UPR-activated caspase, since ER stress disrupts the interactions between TRAF2 and procaspase-12, probably due to P-IRE1 binding to TRAF2, which in turn leads to the conversion of procaspase-12 into the active form [28][29].
Finally, ATF6, upon release from BiP, traffics to the Golgi compartment, where it is cleaved to an active transcription factor (cATF6) by serine protease site-1 (S1P) and metalloprotease site-2 (S2P). The activated ATF6 induces the upregulation of ER stress-responsive genes, including BiP and CHOP, protein folding and ERAD genes. It is also well known that active ATF6 and its transcriptional activity are modulated by p38-dependent phosphorylation [30][31][32] (Figure 1).
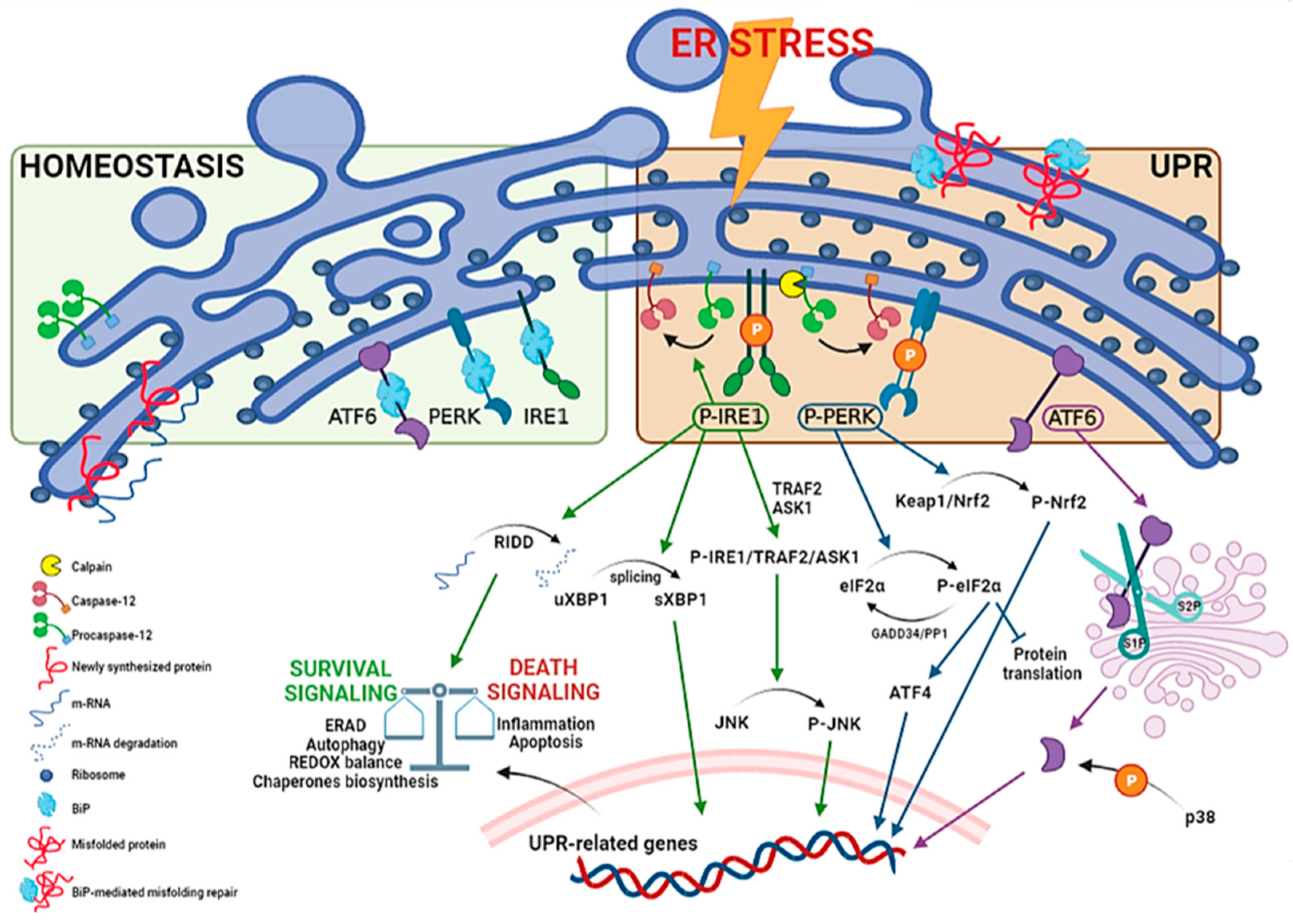
Figure 1. Unfolded protein response pathways. ER stress may be generated by different phenomena that trigger the UPR. The process requires activation by dimerization and autophosphorylation for PERK and IRE1, and intermembrane proteolysis for ATF6 (90 KDa to cleaved form, 50 KDa). Furthermore, UPR activates ER-resident caspase-12. The precise mechanism is not clear, but various processes including interaction with calpain, a calcium-activated cysteine proteases, or activation of the P-IRE1 pathway may contribute to its activation. P-IRE1 induces pro-survival genes to increase protein folding and ERAD. This is allowed through regulated IRE1-dependent decay (RIDD), which degrades several mRNAs, reducing the ER load, as well as IRE1-dependent splicing of uXBP1 to the active spliced factor sXBP1. If ER stress persists, pro-apoptotic genes are activated by a complex of IRE1 with TRAF2 and ASK1, leading to JNK phosphorylation (JNK activation can be distinguished in an early and transient antiapoptotic and a later phase, that coincides with activation of caspases). P-PERK activates, in the short-term, a pro-survival response by eIF2α phosphorylation, leading to the inhibition of protein translation. Furthermore, P-PERK triggers the Nrf2-based antioxidant pathway due to Keap1/Nrf2 complex phosphorylation. Simultaneously, ATF4 transcription leads to pro-survival genes (e.g., ERAD, autophagy pathways). In the long-term, there is the activation of pro-apoptotic genes, such as CHOP, and protein translation reactivation by GADD34/PP1 complex that dephosphorylates P-eIF2α. ATF6 is transported to the cis-Golgi for cleavage by site 1 and site 2 proteases (S1P, S2P), resulting in an active cleaved form (cATF6) that acts as transcription factor to enhance folding and ERAD genes. p38 MAPK may modulate cATF6 activity by phosphorylation.
3. ER Stress in Diseases Pathogenesis
UPR is activated in several disorders, including inflammation, diabetes, metabolic and neurodegenerative diseases, as well as in cancer [33][34][35][36][37][38][39]. UPR can have contrasting effects being either cell protective or cell destructive depending on the strength or duration of the insult [40][41].
It is well known that the UPR pathways are chronically activated in cancer since tumor cells are challenged by microenvironments. Acidosis, hypoxia and hypoglycemia, for instance, lead to the induction of protective module of ER stress. This is supported by the presence of permanently elevated levels of pro-survival GRP78 protein in many tumor cells (Figure 2), exerting protective effects against the cytotoxicity of several chemotherapeutics, such as taxanes, anthracyclines and imidazotetrazines [42][43][44]. On the contrary, CHOP, showing opposite functions than GRP78, generally is not significantly expressed in tumors, despite low and chronic ER stress levels, because the pro-survival action prevails and GRP78 inactivates the ER transmembrane signaling components PERK, IRE1 and ATF6 [45].
Progression of type 2 diabetes requires an increase in pancreatic β-cells for insulin production in order to attenuate insulin resistance. This causes an augmented processing of proinsulin to insulin in the ER that, combined with an enhancement of free fatty acids and glucose, triggers chronic ER stress. If these conditions are maintained for long-term periods, as for obese patients and people with unbalanced diets, chronic ER stress may lead to β-cell death, thus initiating a vicious circle of exacerbated hyperglycemia. Instead, high glucose levels, compatible with diabetes and obesity, induce PERK and eIF2α phosphorylation, concomitant inhibition of protein synthesis, activation of JNK and ATF6 and overexpression of ATF4 and CHOP in β-cells (Figure 2). Moreover, recent evidence suggests that free fatty acids, in particular palmitate, activate the ER stress response in vitro [46][47].
During inflammatory response, ROS production notably increases, impairing ER redox balance and disulfide bond formation. It has been shown that extracellular sources of ROS can induce ER stress, through depletion of Ca2+ in the ER. Moreover, ROS can also promote NLRP3 (NLR family, pyrin domain containing 3) inflammasome activation via RIDD (IRE1 pathway) and/or transcription of the activating transcription factor 5 (ATF5) (PERK-eIF2α pathway) via thioredoxin-interacting protein (TXNIP) activation. TXNIP is normally untranslated due to miR-17, which is degraded by RIDD under ER stress conditions. Additionally, ROS trigger inflammatory responses by enhancing the phosphorylation of IκB to induce nuclear factor-κB (NF-κB) signaling by the interaction of IRE1 with TRAF2, phosphorylation of AKT by cleaved ATF6 or translation attenuation by PERK-dependent phosphorylation of eIF2α. The process leads to a decreased expression of both IκB and NF-κB. However, given the shorter half-life of IκB, the resulting increase in NF-κB/IκB ratio prompts inflammation [48][49][50][51] (Figure 2).
UPR signaling also plays a role in neuronal physio-pathological processes and can influence higher brain functions. Accordingly, the accumulation of misfolded proteins is a typical occurrence in many neurodegenerative disorders [52].
Degeneration of substantia nigra dopaminergic neurons and the presence of α-synuclein containing Lewy bodies are peculiar to Parkinson’s disease (PD). α-synuclein is expressed in synaptic vesicles and nervous cell membranes; alterations in α-synuclein post-translational modifications are associated to missense mutations causing dominant familial PD. Thus, these aberrations can cause protein misfolding due to the shift from the predominantly α-helix to β-sheet conformation, followed by plaque formation [53]. For instance, the A53T mutation is related to UPR activation as shown by increased expression of BiP, CHOP and P-eIF2α. Inhibition of eIF2α phosphorylation rescues the A53T cells from apoptosis, suggesting that the activated UPR may promote cell death in these disorders [54] (Figure 2). Further in vitro evidence displays pro-survival effect of cocoa procyanidins on PD model induced by 6-hydroxydopamine inhibiting UPR via PERK phosphorylation block [36].
Alzheimer’s disease (AD) is an age-associated dementia disorder characterized by the accumulation of extracellular β-amyloid (Aβ) peptides and hyperphosphorylated tau (pTau) protein, leading to intracellular protein aggregation in senile plaques. Ca2+ homeostasis is essential to regulate functions of ER chaperones and protein folding. Alterations in Ca2+ balance lead to reduced chaperone activity, protein misfolding and initiation of the UPR. Aβ peptides have been shown to cause severe depletion of ER Ca2+ storage by triggering release into the cytosol [55][56] (Figure 2).
Amyotrophic lateral sclerosis (ALS) represents a progressive neurological disease that primarily affects motoneurons. Bunina bodies, deposition of ubiquitylated proteins and aggregates of mutant superoxide dismutase 1 (mSOD1) and protein disulfide isomerase (PDI) are prodromic of the disease. Contrasting data suggest that UPR activation may cause ER stress-induced apoptosis or may be neuroprotective by strongly triggering autophagy [57][58] (Figure 2). Therefore, it is evident that cellular mechanisms affecting the balance between the protective and apoptotic response of the UPR are critical in these pathologies.
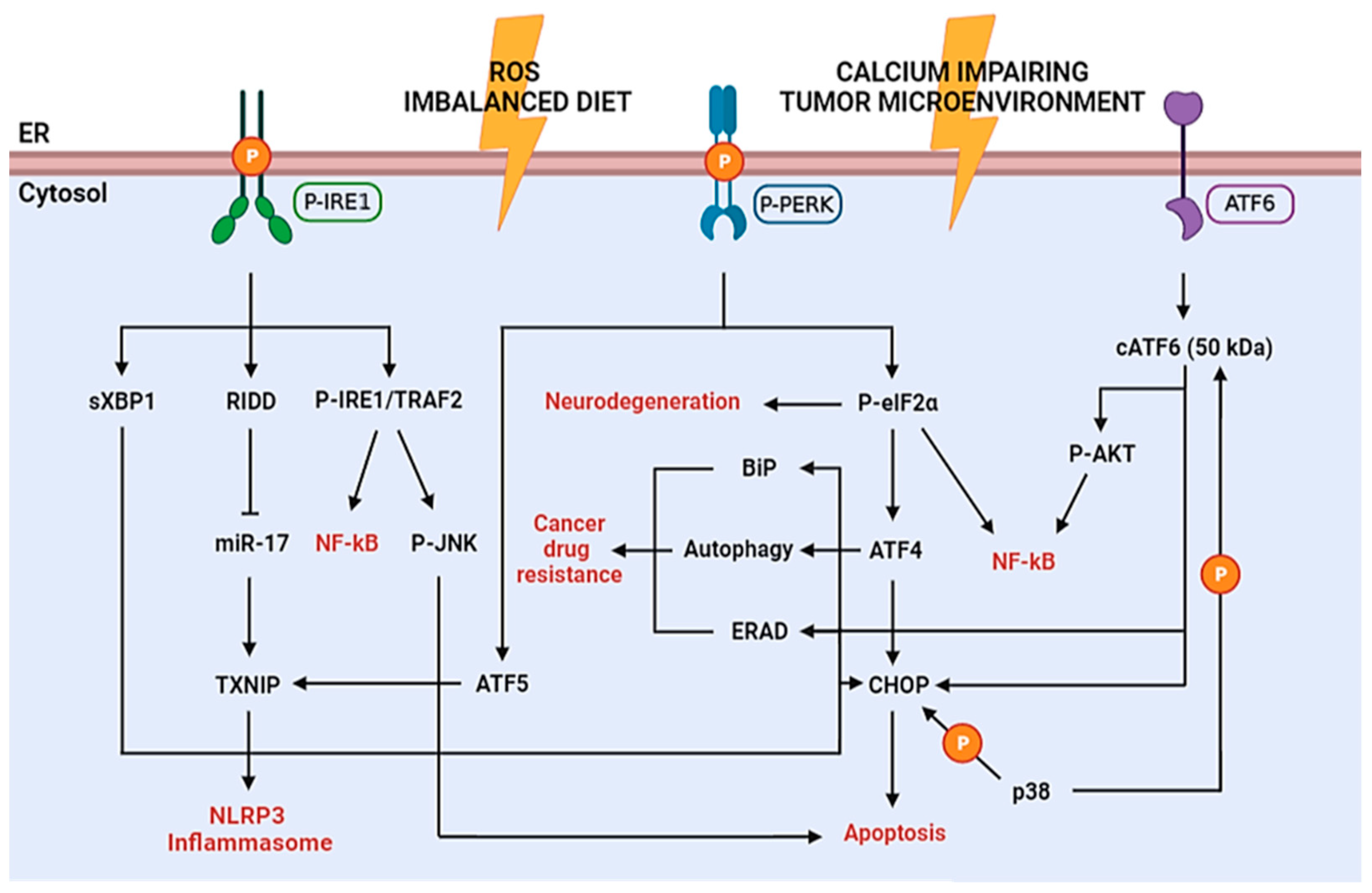
Figure 2. Schematic diagram of the UPR signaling in diseases. P-IRE1 and subsequent sXBP1 directly binds the promoters of UPR genes, which may lead to cell death (CHOP) or survival (ERAD, autophagy, BiP) depending on the intensity of the ER stress. IRE1 activates RIDD-dependent degradation of miR-17, which in normal conditions represses TXNIP, thus enabling increased TXNIP levels and NLRP3 inflammasome activation with consequent inflammatory cytokines production. Furthermore, TXNIP can be induced through the P-PERK–ATF5 pathway to induce inflammasome activation. Moreover, P-IRE1 phosphorylates TRAF2 leading to NF-kB activation (inflammation pathway) or JNK activation (apoptosis pathway). P-PERK phosphorylates eIF2α stopping the protein synthesis. This causes suppression of the synthesis of essential synaptic proteins, leading to neurodegeneration, and inflammation pathway through NF-kB activation. On the other hand, ATF4 expression enhances; this transcription factor represents a double-edged sword since it may activate apoptotic genes (e.g., CHOP, GADD34, caspases), but also pro-survival genes (e.g., BiP and other chaperones, SEL1L, autophagy genes) causing cancer drug resistance in tumor cells. The cleaved form of ATF6 (cATF6) may activate pro-survival or apoptotic response and inflammation pathway (NF-kB), similarly to ATF4. Furthermore, p38 can also phosphorylate CHOP and cATF6 to enhance their activity.
References
- Mickle, A.D.; Shepherd, A.J.; Mohapatra, D.P. Nociceptive TRP Channels: Sensory Detectors and Transducers in Multiple Pain Pathologies. Pharmaceuticals 2016, 9, 72.
- Kim, S.R.; Lee, D.Y.; Chung, E.S.; Oh, U.T.; Kim, S.U.; Jin, B.K. Transient receptor potential vanilloid subtype 1 mediates cell death of mesencephalic dopaminergic neurons in vivo and in vitro. J. Neurosci. 2005, 25, 662–671.
- Andersson, K.E.; Gratzke, C.; Hedlund, P. The role of the transient receptor potential (TRP) superfamily of cation-selective channels in the management of the overactive bladder. BJU Int. 2010, 106, 1114–1127.
- Yang, D.; Kim, J. Emerging role of transient receptor potential (TRP) channels in cancer progression. BMB Rep. 2020, 53, 125–132.
- Phillips, M.; Voeltz, G. Structure and function of ER membrane contact sites with other organelles. Nat. Rev. Mol. Cell Biol. 2016, 17, 69–82.
- Amodio, G.; Pagliara, V.; Moltedo, O.; Remondelli, P. Structural and Functional Significance of the Endoplasmic Reticulum Unfolded Protein Response Transducers and Chaperones at the Mitochondria-ER Contacts: A Cancer Perspective. Front. Cell Dev. Biol. 2021, 26, 641194.
- Shore, G.C.; Papa, F.R.; Oakes, S.A. Signaling cell death from the endoplasmic reticulum stress response. Curr. Opin. Cell Biol. 2011, 23, 143–149.
- Kadowaki, H.; Nishitoh, H. Signaling pathways from the endoplasmic reticulum and their roles in disease. Genes 2013, 4, 306–333.
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 1, 421–438.
- Kitamura, M. Endoplasmic reticulum stress and unfolded protein response in renal pathophysiology: Janus faces. Am. J. Physiol. Renal Physiol. 2008, 295, F323–F334.
- Bhattarai, K.R.; Riaz, T.A.; Kim, H.R.; Chae, H.G. The aftermath of the interplay between the endoplasmic reticulum stress response and redox signaling. Exp. Mol. Med. 2021, 53, 151–167.
- Lindholm, D.; Korhonen, L.; Eriksson, O.; Kõks, S. Recent Insights into the Role of Unfolded Protein Response in ER Stress in Health and Disease. Front. Cell Dev. Biol. 2017, 10, 48.
- Wang, S.; Kaufman, R.J. The impact of the unfolded protein response on human disease. J. Cell Biol. 2012, 25, 857–867.
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332.
- Valenzuela, V.; Collyer, E.; Armentano, D.; Parsons, G.B.; Court, F.A.; Hetz, C. Activation of the unfolded protein response enhances motor recovery after spinal cord injury. Cell Death Dis. 2012, 16, e272.
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell 2002, 3, 99–111.
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J. Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol. 2003, 23, 7198–7209.
- Amodio, G.; Moltedo, O.; Faraonio, R.; Remondelli, P. Targeting the endoplasmic reticulum unfolded protein response to counteract the oxidative stress-Induced endothelial dysfunction. Oxid. Med. Cell. Longev. 2018, 2018, 4946289.
- Amodio, G.; Moltedo, O.; Fasano, D.; Zerillo, L.; Oliveti, M.; Di Pietro, P.; Faraonio, R.; Barone, P.; Pellecchia, M.T.; De Rosa, A.; et al. PERK-Mediated Unfolded Protein Response Activation and Oxidative Stress in PARK20 Fibroblasts. Front. Neurosci. 2019, 27, 673.
- Vattem, K.M.; Wek, R.C. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11269.
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2α. J. Cell. Biol. 2001, 153, 1011–1021.
- Shacham, T.; Patel, C.; Lederkremer, G.Z. PERK Pathway and Neurodegenerative Disease: To Inhibit or to Activate? Biomolecules 2021, 11, 354.
- Maytin, E.V.; Ubeda, M.; Lin, J.C.; Habener, J.F. Stress-inducible transcription factor CHOP/gadd153 induces apoptosis in mammalian cells via p38 kinase-dependent and -independent mechanisms. Exp. Cell Res. 2001, 267, 193–204.
- Vidal, R.L.; Figueroa, A.; Court, F.A.; Thielen, P.; Molina, C.; Wirth, C.; Caballero, B.; Kiffin, R.; Segura-Aguilar, J.; Cuervo, A.M.; et al. Targeting the UPR transcription factor XBP1 protects against Huntington’s disease through the regulation of FoxO1 and autophagy. Hum. Mol. Genet. 2012, 21, 2245–2262.
- Lin, J.H.; Walter, P.; Yen, T.S. Endoplasmic reticulum stress in disease pathogenesis. Annu. Rev. Pathol. 2008, 3, 399–425.
- Bashir, S.; Banday, M.; Qadri, O.; Bashir, A.; Hilal, N.; Nida, I.F.; Rader, S.; Fazili, K.M. The molecular mechanism and functional diversity of UPR signaling sensor IRE1. Life Sci. 2021, 265, 118740.
- Chen, L.; Xu, S.; Liu, L.; Wen, X.; Xu, Y.; Chen, J.; Teng, J. Cab45S inhibits the ER stress-induced IRE1-JNK pathway and apoptosis via GRP78/BiP. Cell Death Dis. 2014, 8, e1219.
- Yoneda, T.; Imaizumi, K.; Oono, K.; Yui, D.; Gomi, F.; Katayama, T.; Tohyama, M. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J. Biol. Chem. 2001, 27, 13935–13940.
- Hitomi, J.; Katayama, T.; Taniguchi, M.; Honda, A.; Imaizumi, K.; Tohyama, M. Apoptosis induced by endoplasmic reticulum stress depends on activation of caspase-3 via caspase-12. Neurosci. Lett. 2004, 4, 27–30.
- Schindler, A.J.; Schekman, R. In vitro reconstitution of ER-stress induced ATF6 transport in COPII vesicles. Proc. Natl. Acad. Sci. USA 2009, 20, 17775–17780.
- Darling, N.J.; Cook, S.J. The role of MAPK signalling pathways in the response to endoplasmic reticulum stress. Biochim. Biophys. Acta 2014, 1843, 2150–2163.
- Kopp, M.C.; Larburu, N.; Durairaj, V.; Adams, C.J.; Ali, M.M.U. UPR proteins IRE1 and PERK switch BiP from chaperone to ER stress sensor. Nat. Struct. Mol. Biol. 2019, 26, 1053–1062.
- Deécio, L.E.; Ales, K.C.; Miriam, C. The Role for Endoplasmic Reticulum Stress in Diabetes Mellitus. Endocr. Rev. 2008, 29, 42–61.
- Chipurupalli, S.; Samavedam, U.; Robinson, N. Crosstalk Between ER Stress, Autophagy and Inflammation. Front. Med. 2021, 5, 758311.
- Madden, E.; Logue, S.E.; Healy, S.J.; Manie, S.; Samali, A. The role of the unfolded protein response in cancer progression: From oncogenesis to chemoresistance. Biol. Cell. 2019, 111, 11–17.
- Vestuto, V.; Amodio, G.; Pepe, G.; Basilicata, M.G.; Belvedere, R.; Napolitano, E.; Guarnieri, D.; Pagliara, V.; Paladino, S.; Rodriquez, M.; et al. Cocoa Extract Provides Protection against 6-OHDA Toxicity in SH-SY5Y Dopaminergic Neurons by Targeting PERK. Biomedicines 2022, 10, 2009.
- Asik, R.M.; Suganthy, N.; Aarifa, M.A.; Kumar, A.; Szigeti, K.; Mathe, D.; Gulyás, B.; Archunan, G.; Padmanabhan, P. Alzheimer’s Disease: A Molecular View of β-Amyloid Induced Morbific Events. Biomedicines 2021, 9, 1126.
- Benedetti, R.; Montani, M.S.G.; Romeo, M.A.; Arena, A.; Santarelli, R.; D’Orazi, G.; Cirone, M. Role of UPR Sensor Activation in Cell Death–Survival Decision of Colon Cancer Cells Stressed by DPE Treatment. Biomedicines 2021, 9, 1262.
- Ciccarese, F. Cancer Metabolism and Resistance to Cell Death: Novel Therapeutic Perspectives. Biomedicines 2022, 10, 1828.
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell. Biol. 2011, 13, 184–190.
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470.
- Lee, A.S. GRP78 induction in cancer: Therapeutic and prognostic implications. Cancer Res. 2007, 15, 3496–3499.
- Lee, E.; Nichols, P.; Spicer, D.; Groshen, S.; Yu, M.C.; Lee, A.S. GRP78 as a novel predictor of responsiveness to chemotherapy in breast cancer. Cancer Res. 2006, 15, 7849–7853.
- Pyrko, P.; Schönthal, A.H.; Hofman, F.M.; Chen, T.C.; Lee, A.S. The unfolded protein response regulator GRP78/BiP as a novel target for increasing chemosensitivity in malignant gliomas. Cancer Res. 2007, 15, 9809–9816.
- Schönthal, A.H. Endoplasmic reticulum stress: Its role in disease and novel prospects for therapy. Scientifica 2012, 2012, 857516.
- Kuo, T.F.; Tatsukawa, H.; Matsuura, T.; Nagatsuma, K.; Hirose, S.; Kojima, S. Free fatty acids induce transglutaminase 2-dependent apoptosis in hepatocytes via ER stress-stimulated PERK pathways. J. Cell. Phys. 2012, 227, 1130–1137.
- Laybutt, D.R.; Preston, A.M.; Akerfeldt, M.C.; Kench, J.G.; Busch, A.K.; Biankin, A.V.; Biden, T.J. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia 2007, 50, 752–763.
- Nathan, C.; Cunningham-Bussel, A. Beyond oxidative stress: An immunologist’s guide to reactive oxygen species. Nat. Rev. Immunol. 2013, 1, 349–361.
- Lerner, A.G.; Upton, J.P.; Praveen, P.V.; Ghosh, R.; Nakagawa, Y.; Igbaria, A.; Shen, S.; Nguyen, V.; Backes, B.J.; Heiman, M.; et al. IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 2012, 16, 250–264.
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225.
- Grootjans, J.; Kaser, A.; Kaufman, R.J.; Blumberg, R.S. The unfolded protein response in immunity and inflammation. Nat. Rev. Immunol. 2016, 16, 469–484.
- Doyle, K.M.; Kennedy, D.; Gorman, A.M.; Gupta, S.; Healy, S.J.; Samali, A. Unfolded proteins and endoplasmic reticulum stress in neurodegenerative disorders. J. Cell. Mol. Med. 2011, 15, 2025–2039.
- Brundin, P.; Li, J.Y.; Holton, J.L.; Lindvall, O.; Revesz, T. Research in motion: The enigma of Parkinson’s disease pathology spread. Nat. Rev. Neurosci. 2008, 9, 741–745.
- Smith, W.W.; Jiang, H.; Pei, Z.; Tanaka, Y.; Morita, H.; Sawa, A.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum. Mol. Genet. 2005, 15, 3801–3811.
- Chami, M.; Checler, F. Alterations of the Endoplasmic Reticulum (ER) Calcium Signaling Molecular Components in Alzheimer’s Disease. Cells 2020, 1, 2577.
- Leissring, M.A.; LaFerla, F.M.; Callamaras, N.; Parker, I. Subcellular mechanisms of presenilin-mediated enhancement of calcium signaling. Neurobiol. Dis. 2001, 8, 469–478.
- Atkin, J.D.; Farg, M.A.; Walker, A.K.; McLean, C.; Tomas, D.; Horne, M.K. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol. Dis. 2008, 30, 400–407.
- Hetz, C.; Glimcher, L.H. Fine-tuning of the unfolded protein response: Assembling the IRE1alpha interactome. Mol. Cell 2009, 11, 551–561.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
958
Revisions:
2 times
(View History)
Update Date:
27 Dec 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No