Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Nuno Silva | -- | 3145 | 2022-12-22 16:00:38 | | | |
| 2 | Sirius Huang | Meta information modification | 3145 | 2022-12-23 04:25:11 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Lima, R.; Monteiro, A.; Salgado, A.J.; Monteiro, S.; Silva, N.A. Pathophysiology of Spinal Cord Injury. Encyclopedia. Available online: https://encyclopedia.pub/entry/39106 (accessed on 10 August 2026).
Lima R, Monteiro A, Salgado AJ, Monteiro S, Silva NA. Pathophysiology of Spinal Cord Injury. Encyclopedia. Available at: https://encyclopedia.pub/entry/39106. Accessed August 10, 2026.
Lima, Rui, Andreia Monteiro, António J. Salgado, Susana Monteiro, Nuno A. Silva. "Pathophysiology of Spinal Cord Injury" Encyclopedia, https://encyclopedia.pub/entry/39106 (accessed August 10, 2026).
Lima, R., Monteiro, A., Salgado, A.J., Monteiro, S., & Silva, N.A. (2022, December 22). Pathophysiology of Spinal Cord Injury. In Encyclopedia. https://encyclopedia.pub/entry/39106
Lima, Rui, et al. "Pathophysiology of Spinal Cord Injury." Encyclopedia. Web. 22 December, 2022.
Copy Citation
Spinal cord injury (SCI) is a disabling condition that disrupts motor, sensory, and autonomic functions. The lack of effective therapeutic strategies for patients with SCI reflects its complex pathophysiology that leads to the point of no return in its function repair and regeneration capacity. Herein, a detailed description of the physiology and anatomy of the spinal cord and the pathophysiology of SCI is presented.
spinal cord injury
pathophysiology
spinal cord
1. Spinal Cord Anatomy and Physiology
The spinal cord is the major communication channel between the body and the brain. Additionally, the spinal cord can independently respond to sensory information without input from the brain through reflex arcs and produce repetitive patterns of motor behavior using self-containing circuits known as Central Pattern Generators (CPGs). The spinal cord extends from the base of the brain in the medulla oblongata through the foremen magnum of the skull to the L1/L2 lumbar vertebra, where it terminates as the conus medullaris. Distal to this end of the spinal cord is a collection of nerve roots called the cauda equina. Like the brain, which is protected by the cranium, the spinal cord is likewise protected by a bone structure called vertebral column. The spinal cord is also protected by three membranes of connective tissue called meninges (dura mater, arachnoid mater, and pia mater). The subarachnoid space (between arachnoid and pia), which is filled with cerebrospinal fluid, and the epidural space (between dura and periosteum), which is filled with loose fibrous and adipose connective tissues, also helps to protect the spinal cord [1][2][3].
Contrary to the brain, the spinal cord’s gray matter is centrally surrounded by white matter. The gray matter comprises neuronal cell bodies, interneurons, dendrites of efferent neurons, entering fibers of afferent neurons, neuroglia cells, and axons which are predominantly unmyelinated. Unlike gray matter, white matter is a collection of interconnecting fibers composed mostly of myelinated sensory and motor axons. The surrounding white matter is composed mostly of groups of myelinated axons [1][3][4].
The spinal cord has numerous groups of nerve fibers going towards and coming from the brain. The tracts are described according to the funiculus within which they are located. The ascending tracts usually start with the prefix spino- and end with the name of the brain region where spinal cord fibers first synapse (e.g., spinothalamic tract). The descending motor tracts begin with the prefix denoting the brain region that gives rise to the fibers and ends with the suffix -spinal (e.g., corticospinal tract) [1][3][5].
The spinal nerves leave and enter the spinal cord by the ventrolateral and dorsolateral sulcus. Sensory neurons enter the spinal cord by the dorsal side, while axons of efferent (motor) neurons leave the spinal cord on the ventral side via the ventral roots. Near the cord, the dorsal and ventral roots from the same level combine to form a spinal nerve on each side of the spinal cord. The spinal nerves’ nomenclature depends on the vertebral levels from which they exit: cervical (C1–C8), thoracic (T1–T12), lumbar (L1–L5), sacral (S1–S5), and coccygeal (Co1). Cervical nerves are responsible for controlling the muscles and glands, and receive sensory input from the neck, shoulder, arm and hand. Thoracic nerves are associated with the chest and abdominal walls, and the lumbar nerves carry information related to the hip and leg. The sacral nerves are associated with the genitals and lower digestive tract control, and, finally, the coccygeal nerve supplies the skin over the coccyx [1][3].
2. Spinal Cord Injury
A spinal cord injury is a devastating event that leads to motor, sensory, and autonomic dysfunctions. The complexity of this event and the lack of an effective treatment make SCI a worldwide problem. A study performed by the Global Burden of Diseases, Injuries and Risk Factors (GBD) reported 0.93 million (0.78–1.16 million) new SCI cases globally, with a prevalence of 27.04 million cases (24.98–30.15 million) [6]. The annual incidence of SCI varies greatly from region to region: 7 to 37 cases per 100,000 individuals [6]. In the United States, traffic accidents are currently the leading cause of injury (38%), followed by falls (30%), violence (13.5%), and sports/recreation accidents (9%) [7]. The average age at injury is 42 years, and 80% of spinal cord injuries occur in males [7].
The American Spinal Cord Injury Association (ASIA) Impairment Scale defines the degree of neurological loss as: A (complete sensorimotor loss below the lesion including absent sacral sensation), B (sensory but no motor function below the lesion level), C (some motor preservation, but the majority of muscles are less than 3), and D (muscle grade is 3 or greater in the majority of groups below the lesion) [8]. Incomplete tetraplegia is currently the most frequent injury (45%), followed by incomplete paraplegia (21.3%), complete paraplegia (20%), and complete tetraplegia (13.3%) [7]. Less than 1% of persons experience complete recovery by hospital discharge [7].
2.1. Primary Injury
SCI results from an insult that damages the spinal cord, which can be subdivided into non-traumatic and traumatic. Non-traumatic injury occurs when an acute or chronic disease, such as a tumor, infection, or degenerative disease, causes damage to the spinal cord. The traumatic and most common SCI results from a traumatic impact that fractures or dislocates vertebrae. The initial mechanical impact to the spinal cord at the time of injury is denominated primary injury. The most common form of primary impact is compression injury, which typically occurs through burst fractures, with bone fragments compressing the spinal cord, or through fracture-dislocation injuries [9][10]. Impact alone with transient compression is observed less frequently, but most commonly in hyperextension injuries [9]. Distraction injuries occur when two adjacent vertebrae are pulled apart, causing the spinal column to stretch and tear in the axial plane [9]. Lastly, laceration and transection injuries can occur through projectile injuries, severe dislocations, or sharp bone fragment dislocations, and can present high variability from minor injuries to complete transection [9]. Regardless of the form of primary injury, these forces will directly damage the neurons, glial cells, and the neurovasculature of the spinal cord [9]. Overall, the extent of the primary injury and its level determines the severity and outcome of SCI [11].
2.2. Secondary Injury
Following the primary injury, a derived degenerative process initiates within minutes and hours, which is proportional to the magnitude of the initial insult. This resultant process is commonly denominated by secondary injury. This comprises permeability and vascular alterations, ionic disruption and glutamate excitotoxicity, metabolic alterations, a dysfunctional inflammatory response, and initiation of glial scarring [12]. The main tissue alterations promoted by the secondary injury cascade of events are described below.
2.2.1. Permeability and Vascular Alterations
The hemorrhage associated with the “primary injury,” coupled with systemic hypotension, culminates in a major reduction in the blood flow at the lesion site [13]. Over time, the decreased blood flow leads to ischemia. Although it remains unclear, the retraction of the blood supply may be due to microvascular detriment, hypotension, loss of autoregulation, and an increase in interstitial pressure [14]. Ultimately, cells are deprived of oxygen and glucose, leading to necrosis [15]. Moreover, post-injury hemorrhage and ischemia also impact blood–spinal cord barrier (BSCB) permeability. The direct alteration of BSCB endothelial cells promotes the infiltration of immune mediators that leads to edema, and might increase the pro-inflammatory environment in the injured spinal cord [16].
2.2.2. Ionic Disruption and Glutamate Excitotoxicity
After the insult, the homeostatic ionic balance is severely compromised. Membrane depolarization and ATPase disruption enhance neuronal and glial cell death by increasing intracellular calcium (Ca2+) levels. Additionally, there is an exacerbated release of glutamate to the extracellular space, reaching neurotoxic levels [17]. Glutamate is a well-described excitatory neurotransmitter, regulated by Ca2+ flux at the synaptic cleft. After SCI, there is an excessive release of this amino acid [14][17], and consequently, excessive activation of glutamate receptors (NMDA and AMPA) that leads to an increase in sodium (Na+) and Ca2+ influx. Ionic dysregulation directly impacts neuronal and glial cells, especially oligodendrocytes and neurons, leaving them vulnerable to cell death. In addition, axonal degeneration is mediated by Ca2+ influx from the endoplasmic reticulum (ER) through the inositol triphosphate (IP3) receptor, which promotes mitochondrial permeability [18]. Overall, glutamate excitotoxicity disturbs ionic homeostasis and normal mitochondrial functioning, resulting in axonal demyelination and neuronal loss at the injury site [12][19].
2.2.3. Metabolic Alterations
Ischemia, oxygen deprivation, and oxidative stress lead to the production of high levels of reactive oxygen species (ROS) and reactive nitrogen species (RNS) [14][20]. As a consequence, ROS and RNS are strongly reactive with polyunsaturated fatty acid of the cellular membrane, leading not only to lipid peroxidation, but also to damage at the protein and nucleic acid levels. Furthermore, the formation of free radicals also invokes architectonic alterations of the cytoskeleton and organelle membranes, mitochondrial dysfunction, and increased intracellular Ca2+ uptake [9][14].
The formation of specific aldehyde products promotes cell membrane disruption, affecting nearby healthy cells. Additionally, impairments at the metabolic levels are also observed in the normal functioning of the transmembrane (Na+/K+)-ATPase enzyme. As a major Ca2+ pump, ATPase is crucial for maintaining neuronal excitability and alterations in its activity, triggering axonal and neuronal loss [9][19].
2.2.4. Inflammatory Response
Inflammation is a major “secondary injury” event, and its dysregulated nature leads to more neuronal damage [21]. Initiation of the “secondary injury” leads to cell activation of astrocytes, fibroblasts, pericytes, and microglia. The BSCB disruption further drives injury progression by facilitating the infiltration of non-resident cells. Peripheral immune cells invade the injury site and chronically persist within the spinal cord [22]. Fibroblasts, which infiltrate from the periphery or differentiate from other resident cells, deposit inhibitory extracellular matrix (ECM) components that aggravate the inflammatory environment [23]. Moreover, SCI generates cellular debris and releases intracellular proteins that induce potent inflammatory stimuli. This debris signal, also called damage-associated molecular patterns (DAMPs), is usually hidden from immune surveillance within the intact CNS [24][25]. After an injury, DAMPs engage pattern recognition receptors (PRR) of inflammatory cells involved in foreign microbe detection [26]. As a result of the rapid DAMP- and PRR-mediated activation, resident and peripheral inflammatory cells are recruited to the lesion site [24][25]. Consequently, these cells release various oxidative stress regulators, cytokines, chemokines, and other inflammatory mediators that exacerbate the inflammatory response [24][27].
Regarding microglia, the cellular morphology and protein expression profiles are altered following SCI. Microglia cells retract their processes and assume an amoeboid morphology, making them better prepared for phagocytosis and debris clearance. Reactive microglia closely resemble circulating macrophages in terms of morphology, protein expression profile, and function [28]. Together with morphological changes, the release of chemokines and cytokines recruits neutrophils and macrophages into the injured spinal cord [29]. The first type of infiltrating immune cells are the neutrophils, which, in rodents and humans, have their peak within the spinal cord around 1-day post-injury [27][30][31]. The by-products produced after neutrophil-mediated phagocytosis create a cytotoxic environment with the production of ROS and reactive nitrogen species (RNS) [32].
Moreover, neutrophils persist chronically at low levels in the spinal cord, but decrease within a week of injury in both rodents and humans [22][33]. Monocyte-derived macrophages also infiltrate the spinal cord [34] and contribute, along with proteolytic enzymes, ROS, and inflammatory cytokines, to the injury microenvironment. They also perform critical functions, such as debris clearance, cellular remodeling, and production of pro-regenerative factors [28][35]. Likewise, CNS reactive B- and T-cells also infiltrate the spinal cord, and have been suggested to promote axonal injury and demyelination [9]. Recently, single-cell transcriptomic analyses revealed that even in the chronic phase, the major cell types of the spinal cord are still considerably deviated from uninjured states, and that microglia were the most dynamically altering cell population after SCI. Even by day 42, when tissue homeostasis is stabilized, the microglial populations diverge from those before the injury, indicating the long-lasting alterations in the immune microenvironment after injury [36]. It is important to point out, as the more we understand the practical role of the inflammatory response, the more obvious it becomes that this response can support both beneficial and detrimental effects on recovery. Indeed, an acute inflammatory response is needed and crucial for successfully repairing the injured tissue [37]; however, after this initial response, it is important to resolve the inflammatory response by the complex orchestration of different cells and molecular events [38].
2.2.5. Inhibitory Environment
The regeneration of CNS following injury is reduced due to multiple inhibitory factors at the injury site. Several researchers have shown that there is an initial growth response following injury; however, once axons encounter this inhibitory environment, the growth is blocked, leaving dystrophic axonal end bulbs in their place [39]. Within the CNS, cells are surrounded by an ECM composed of a complex and interactive network of glycoproteins, proteoglycans, and glycosaminoglycans [40]. Under different circumstances, these molecules can either promote neurite outgrowth, such as during neuronal development [41], or inhibit it, such as after injury [42] or after neural degeneration [43].
Axonal retraction occurs in two phases: an early axon intrinsic, cytoskeleton-associated phase, in which Ca2+-dependent activation of calpain proteases leads to cytoskeletal breakdown [44], and a macrophage-dependent phase, in which infiltration of phagocytic macrophages correlates with retraction of dystrophic axons [45]. Alongside this, there is an increase in the number of inhibitory proteins, including myelin-associated inhibitors (MAIs), chondroitin sulfate proteoglycans (CSPGs), as well as growth-inhibiting molecules such as proneurotrophins [46].
Nogo-A, oligodendrocytes myelin glycoprotein (OMgp), and myelin-associated glycoprotein (MAG) have all been identified as MAIs that can collapse axonal growth cones and inhibit neurite outgrowth [47]. Nogo-A was identified as a neurite growth inhibitor in the 1980s [48]. The evidence of inhibitory effects of Nogo-A came from in vitro studies in which exposure of chicken retinal ganglion and rat dorsal root ganglion (DRG) neurons to Nogo-A was shown to inhibit neurite outgrowth and induce growth cone collapse [49][50]. The OMgp is also expressed in oligodendrocytes and several types of CNS neurons, such as pyramidal cells in the hippocampus and Purkinje cells in the cerebellum, among others [51]. Although less is known about OMgp in comparison to Nogo-A and MAG, it has also been shown to be a potent inhibitor of neurite outgrowth in multiple cell lines and primary neuronal cultures [52][53].
MAG is a minor component of mature, compact myelin, enriched in the periaxonal membrane of the myelin sheath, and is expressed by oligodendrocytes and Schwann cells [54]. The inhibitory effect of MAG was found in studies investigating its interaction with primary neurons. Purified recombinant MAG was found to block neurite outgrowth and induce growth cone retraction [55][56]. The inhibitory properties of MAG were further confirmed by Tang and colleagues, demonstrating that myelin from MAG knockout mice was not inhibitory to the growth of DRG neurons in vitro compared to myelin from wild-type mice [57]. Furthermore, inhibition of neurite outgrowth was completely abolished by immunodepletion of MAG from the soluble fraction of myelin-conditioned media [58]. These observations suggest that soluble MAIs, likely released after injury, can influence the growth capacity of neurons and axons in addition to myelin debris.
The ECM of the CNS is rich in CSPGs, some existing within the extracellular milieu and others associated with specific structures. Within the CNS, CSPGs can associate with specialized structures, denominated perineuronal nets (PNNs), which surround the soma and dendrites of mature neurons. The PNNs are ECM proteins including hyaluronan, CSPGs, and linking proteins [59]. There are also a number of CSPGs, such as brevican, neurocan, aggrecan, and versican, which bind to the hyaluronan backbone of the PNN [59]. Maintenance of this specialized structure is important for synaptic and network stabilization and homeostasis. Specifically, PNNs stabilize mature neurons by reducing dendritic spine plasticity [60], forming a scaffold for synaptic inhibitory molecules [61] and also restricting the movement of receptors at the synapse [62]. The formation and maturation of PNNs are concurrent with the development and maturation of the nervous system. After injury, CSPGs are actively secreted into the ECM, mainly by reactive astrocytes [63], but with a minor component secretion by macrophages and oligodendrocytes [64][65][66]. This results in an abundance of CSPGs at the injury site, adding to the inhibitory milieu. The inhibitory effect of the CSPGs is mediated through the protein tyrosine phosphatase sigma (PTPσ) receptor. When CSPGs bind to PTPσ receptors, the GTPase Rho/ROCK signaling pathway is activated. In neurons, this inhibits axonal growth, leading the growth cone into a dystrophic state [42][67][68].
2.2.6. Spinal Cord Scarring
As referenced above, SCI activates astrocytes, pericytes, and fibroblasts, promoting the development of a glial/fibrotic scar. Astrocytes activation and subsequent glial scar boundaries are enhanced by the increase in transforming growth factor-beta (TGF-β) [69][70][71]. TGF-β increases microglia/macrophage and astrocyte activation, as well as fibronectin and laminin deposition [70]. Moreover, the signal transducer and activator of the transcription 3 (STAT3) transcription factor is important in establishing glial scar borders that isolate infiltrating cells into the lesion epicenter [72][73].
The deposition of connective tissue and reactive gliosis creates a physical barrier, providing nonspecific topographical cues which affect cellular migration [74][75]. The removal of some inhibitory ECM components, such as CSPGs, improves neurite growth in vivo. However, the removal of other components, such as collagen, fails to promote regeneration and recovery [76]. Together with the chemical components of the scar, stiffness of the newly created ECM also acts as a physical barrier that inhibits axon growth [76][77]. It is important to note that the role of the scar is complex. Some studies have shown the beneficial effects of the glial scar, namely repairing the BSCB, which restrains the inflammatory response, and toxic species sequestration to the injury site [78][79]. Moreover, astrocytes’ capacity to support axon growth and, therefore, neural plasticity in mammalians has been increasingly documented [80]. Additionally, non-mammalian injury models have highlighted glial bridges’ importance for neuronal regeneration [81][82]. In 2017, Hara and coworkers published a study characterizing astrocytes’ varying phenotypes, specifically regarding a lesion site [83]. In this study, three distinct subtypes of astrocytes associated with the glial scar were characterized: naïve, reactive, and scar-forming astrocytes [83]. Interestingly, when the reactive astrocytes were transplanted into the naïve spinal cord, they reverted to naïve astrocytes; likewise, they converted to scar-forming astrocytes when transplanted into an injury site, demonstrating that the environment dictates astrocytic phenotype and consequently glial scar-mediated inhibition [83]. Given the diversity of astrocytes, the next challenge will be to determine context-dependent astrocyte functions, including their regulation of neuronal repair.
2.3. Chronic Phase
Following the secondary injury, the chronic phase is established, and this can lead to the continuous expansion of the lesion site of the patients with SCI. The chronic phase is characterized by scar maturation, cystic cavitation, and axonal dieback [19][84][85]. The process of Wallerian degeneration of injured axons is ongoing, and it may take years for severed axons and their cell bodies to be entirely removed [86]. The lesion may not remain static and syrinx formation may occur, commonly causing dissociated sensory reduction, deterioration of motor function, and neuropathic pain [87][88][89][90].
As described, SCI pathophysiology involves many mechanistically distinct processes that interact in order to both limit and enhance recovery following injury (Figure 1). This dichotomy is well demonstrated by the astrocytic response, which serves to reestablish the blood–brain barrier (BBB), restore ionic homeostasis, and limit immune cell infiltration while severely limiting the ability of axons to regenerate and diminishing functional recovery through the formation of the astrocytic scar. An important consideration for understanding the pathophysiology of human SCI is that each injury is unique, both in cause and resultant damage.
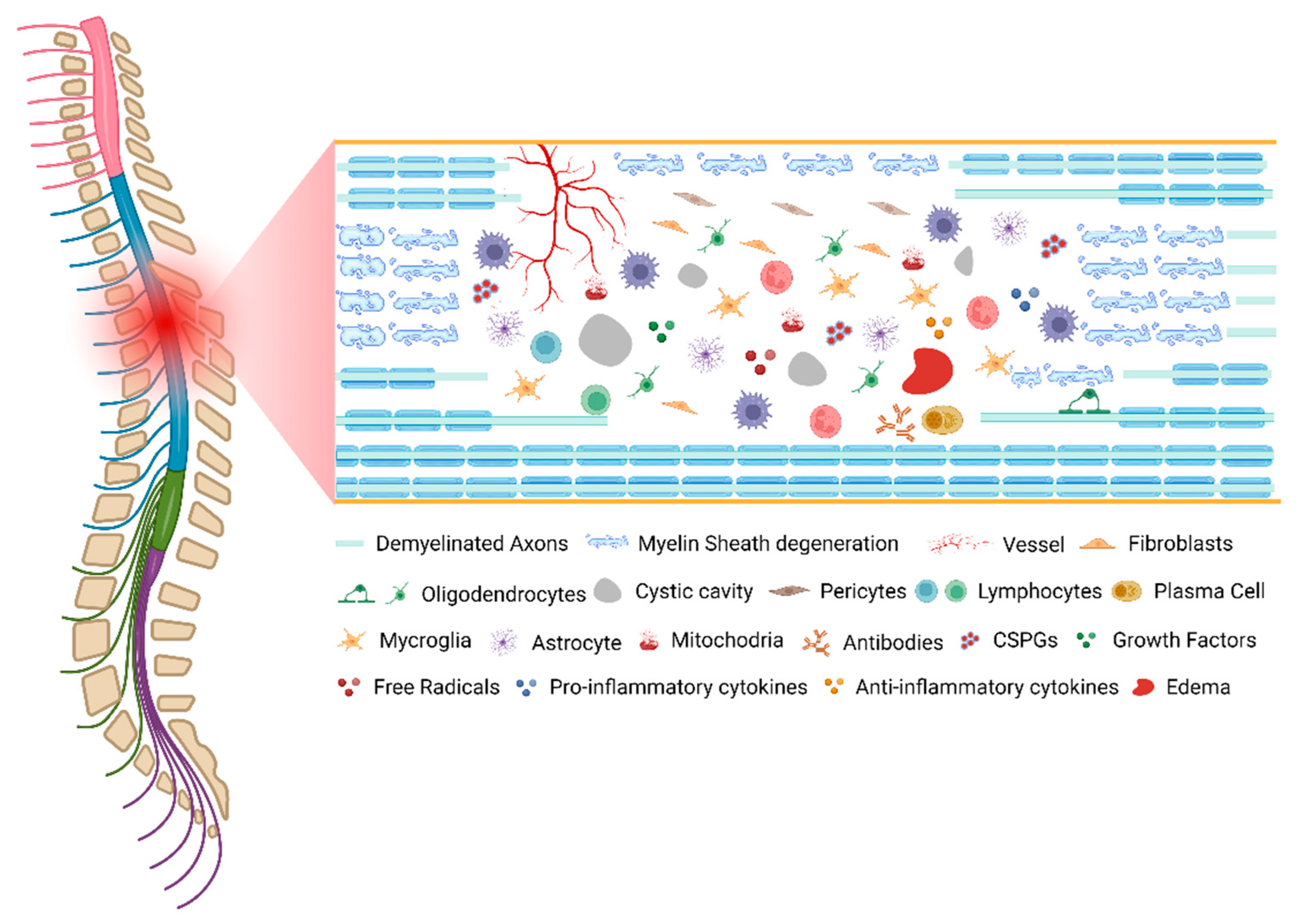
Figure 1. The pathophysiology of spinal cord injury. Created with BioRender.com (accessed on 11 October 2022).
References
- Silva, N.A.; Sousa, N.; Reis, R.L.; Salgado, A.J. From basics to clinical: A comprehensive review on spinal cord injury. Prog. Neurobiol. 2014, 114, 25–57.
- Adigun, O.O.; Reddy, V.; Varacallo, M. Anatomy, Back, Spinal Cord; StatPearls: Treasure Island, Finland, 2022.
- Harrow-Mortelliti, M.; Reddy, V.; Jimsheleishvili, G. Physiology, Spinal Cord; StatPearls: Treasure Island, Finland, 2022.
- Mercadante, A.A.; Tadi, P. Neuroanatomy, Gray Matter; StatPearls: Treasure Island, Finland, 2022.
- Kaiser, J.T.; Lugo-Pico, J.G. Neuroanatomy, Spinal Nerves; StatPearls: Treasure Island, Finland, 2022.
- Injury, G.B.D.T.B.; Injury, C.S.C. Global, regional, and national burden of traumatic brain injury and spinal cord injury, 1990-2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 56–87.
- SCI Facts and Figures. J. Spinal Cord Med. 2017, 40, 126–127.
- Roberts, T.T.; Leonard, G.R.; Cepela, D.J. Classifications In Brief: American Spinal Injury Association (ASIA) Impairment Scale. Clin. Orthop. Relat. Res. 2017, 475, 1499–1504.
- Alizadeh, A.; Dyck, S.M.; Karimi-Abdolrezaee, S. Traumatic Spinal Cord Injury: An Overview of Pathophysiology, Models and Acute Injury Mechanisms. Front. Neurol. 2019, 10, 282.
- Muller-Jensen, L.; Ploner, C.J.; Kroneberg, D.; Schmidt, W.U. Clinical Presentation and Causes of Non-traumatic Spinal Cord Injury: An Observational Study in Emergency Patients. Front. Neurol. 2021, 12, 701927.
- Fehlings, M. Essentials of Spinal Cord Injury: Basic Research to Clinical Practice; Thieme: New York, NY, USA, 2013.
- Hachem, L.D.; Fehlings, M.G. Pathophysiology of Spinal Cord Injury. Neurosurg. Clin. N. Am. 2021, 32, 305–313.
- Hernandez-Gerez, E.; Fleming, I.N.; Parson, S.H. A role for spinal cord hypoxia in neurodegeneration. Cell Death Dis. 2019, 10, 861.
- Quadri, S.A.; Farooqui, M.; Ikram, A.; Zafar, A.; Khan, M.A.; Suriya, S.S.; Claus, C.F.; Fiani, B.; Rahman, M.; Ramachandran, A.; et al. Recent update on basic mechanisms of spinal cord injury. Neurosurg. Rev. 2020, 43, 425–441.
- Anwar, M.A.; Al Shehabi, T.S.; Eid, A.H. Inflammogenesis of Secondary Spinal Cord Injury. Front. Cell. Neurosci. 2016, 10, 98.
- Campos, J.; Silva, N.A.; Salgado, A.J. Nutritional interventions for spinal cord injury: Preclinical efficacy and molecular mechanisms. Nutr. Rev. 2022, 80, 1206–1221.
- Peplow, P.; Dambinova, S.A.; Gennarelli, T.A.; Martinez, B. Acute Brain Impairment: Scientific Discoveries and Translational Research; Royal Society of Chemistry: London, UK, 2018.
- Villegas, R.; Martinez, N.W.; Lillo, J.; Pihan, P.; Hernandez, D.; Twiss, J.L.; Court, F.A. Calcium release from intra-axonal endoplasmic reticulum leads to axon degeneration through mitochondrial dysfunction. J. Neurosci. 2014, 34, 7179–7189.
- Anjum, A.; Yazid, M.D.; Daud, M.F.; Idris, J.; Ng, A.M.H.; Naicker, A.S.; Ismail, O.H.R.; Kumar, R.K.A.; Lokanathan, Y. Spinal Cord Injury: Pathophysiology, Multimolecular Interactions, and Underlying Recovery Mechanisms. Int. J. Mol. Sci. 2020, 21, 7533.
- Liu, D.; Ling, X.; Wen, J.; Liu, J. The role of reactive nitrogen species in secondary spinal cord injury: Formation of nitric oxide, peroxynitrite, and nitrated protein. J. Neurochem. 2000, 75, 2144–2154.
- Monteiro, S.; Pinho, A.G.; Macieira, M.; Serre-Miranda, C.; Cibrao, J.R.; Lima, R.; Soares-Cunha, C.; Vasconcelos, N.L.; Lentilhas-Graca, J.; Duarte-Silva, S.; et al. Splenic sympathetic signaling contributes to acute neutrophil infiltration of the injured spinal cord. J. Neuroinflamm. 2020, 17, 282.
- Beck, K.D.; Nguyen, H.X.; Galvan, M.D.; Salazar, D.L.; Woodruff, T.M.; Anderson, A.J. Quantitative analysis of cellular inflammation after traumatic spinal cord injury: Evidence for a multiphasic inflammatory response in the acute to chronic environment. Brain 2010, 133 Pt 2, 433–447.
- Wang, H.; Xia, Y.; Li, B.; Li, Y.; Fu, C. Reverse Adverse Immune Microenvironments by Biomaterials Enhance the Repair of Spinal Cord Injury. Front. Bioeng. Biotechnol. 2022, 10, 812340.
- Orr, M.B.; Gensel, J.C. Spinal Cord Injury Scarring and Inflammation: Therapies Targeting Glial and Inflammatory Responses. Neurotherapeutics 2018, 15, 541–553.
- Wu, X.; Yan, Y.; Zhang, Q. Neuroinflammation and Modulation Role of Natural Products After Spinal Cord Injury. J. Inflamm. Res. 2021, 14, 5713–5737.
- Kigerl, K.A.; de Rivero Vaccari, J.P.; Dietrich, W.D.; Popovich, P.G.; Keane, R.W. Pattern recognition receptors and central nervous system repair. Exp. Neurol. 2014, 258, 5–16.
- Hellenbrand, D.J.; Quinn, C.M.; Piper, Z.J.; Morehouse, C.N.; Fixel, J.A.; Hanna, A.S. Inflammation after spinal cord injury: A review of the critical timeline of signaling cues and cellular infiltration. J. Neuroinflamm. 2021, 18, 284.
- David, S.; Kroner, A. Repertoire of microglial and macrophage responses after spinal cord injury. Nat. Rev. Neurosci. 2011, 12, 388–399.
- Hao, J.; Li, B.; Duan, H.Q.; Zhao, C.X.; Zhang, Y.; Sun, C.; Pan, B.; Liu, C.; Kong, X.H.; Yao, X.; et al. Mechanisms underlying the promotion of functional recovery by deferoxamine after spinal cord injury in rats. Neural. Regen. Res. 2017, 12, 959–968.
- Zivkovic, S.; Ayazi, M.; Hammel, G.; Ren, Y. For Better or for Worse: A Look Into Neutrophils in Traumatic Spinal Cord Injury. Front. Cell. Neurosci. 2021, 15, 648076.
- Sutherland, T.C.; Mathews, K.J.; Mao, Y.; Nguyen, T.; Gorrie, C.A. Differences in the Cellular Response to Acute Spinal Cord Injury between Developing and Mature Rats Highlights the Potential Significance of the Inflammatory Response. Front. Cell. Neurosci. 2016, 10, 310.
- Soderblom, C.; Lee, D.H.; Dawood, A.; Carballosa, M.; Santamaria, A.J.; Benavides, F.D.; Jergova, S.; Grumbles, R.M.; Thomas, C.K.; Park, K.K.; et al. 3D Imaging of Axons in Transparent Spinal Cords from Rodents and Nonhuman Primates. eNeuro 2015, 2, ENEURO.0001-15.2015.
- Fleming, J.C.; Norenberg, M.D.; Ramsay, D.A.; Dekaban, G.A.; Marcillo, A.E.; Saenz, A.D.; Pasquale-Styles, M.; Dietrich, W.D.; Weaver, L.C. The cellular inflammatory response in human spinal cords after injury. Brain 2006, 129 Pt 12, 3249–3269.
- Donnelly, D.J.; Popovich, P.G. Inflammation and its role in neuroprotection, axonal regeneration and functional recovery after spinal cord injury. Exp. Neurol. 2008, 209, 378–388.
- Gensel, J.C.; Zhang, B. Macrophage activation and its role in repair and pathology after spinal cord injury. Brain Res. 2015, 1619, 1–11.
- Li, C.; Wu, Z.; Zhou, L.; Shao, J.; Hu, X.; Xu, W.; Ren, Y.; Zhu, X.; Ge, W.; Zhang, K.; et al. Temporal and spatial cellular and molecular pathological alterations with single-cell resolution in the adult spinal cord after injury. Signal. Transduct. Target Ther. 2022, 7, 65.
- Bollaerts, I.; Van Houcke, J.; Andries, L.; De Groef, L.; Moons, L. Neuroinflammation as Fuel for Axonal Regeneration in the Injured Vertebrate Central Nervous System. Mediat. Inflamm. 2017, 2017, 9478542.
- Monteiro, S.; Salgado, A.J.; Silva, N.A. Immunomodulation as a neuroprotective strategy after spinal cord injury. Neural. Regen. Res. 2018, 13, 423–424.
- Hill, C.E. A view from the ending: Axonal dieback and regeneration following SCI. Neurosci. Lett. 2017, 652, 11–24.
- Chelyshev, Y.A.; Kabdesh, I.M.; Mukhamedshina, Y.O. Extracellular Matrix in Neural Plasticity and Regeneration. Cell. Mol. Neurobiol. 2022, 42, 647–664.
- Wiese, S.; Faissner, A. The role of extracellular matrix in spinal cord development. Exp. Neurol. 2015, 274 Pt B, 90–99.
- Lang, B.T.; Cregg, J.M.; DePaul, M.A.; Tran, A.P.; Xu, K.; Dyck, S.M.; Madalena, K.M.; Brown, B.P.; Weng, Y.L.; Li, S.; et al. Modulation of the proteoglycan receptor PTPsigma promotes recovery after spinal cord injury. Nature 2015, 518, 404–408.
- Song, I.; Dityatev, A. Crosstalk between glia, extracellular matrix and neurons. Brain Res. Bull. 2018, 136, 101–108.
- Kerschensteiner, M.; Schwab, M.E.; Lichtman, J.W.; Misgeld, T. In vivo imaging of axonal degeneration and regeneration in the injured spinal cord. Nat. Med. 2005, 11, 572–577.
- Busch, S.A.; Horn, K.P.; Silver, D.J.; Silver, J. Overcoming macrophage-mediated axonal dieback following CNS injury. J. Neurosci. 2009, 29, 9967–9976.
- Fan, B.; Wei, Z.; Yao, X.; Shi, G.; Cheng, X.; Zhou, X.; Zhou, H.; Ning, G.; Kong, X.; Feng, S. Microenvironment Imbalance of Spinal Cord Injury. Cell Transplant. 2018, 27, 853–866.
- Boghdadi, A.G.; Teo, L.; Bourne, J.A. The Involvement of the Myelin-Associated Inhibitors and Their Receptors in CNS Plasticity and Injury. Mol. Neurobiol. 2018, 55, 1831–1846.
- Caroni, P.; Savio, T.; Schwab, M.E. Central nervous system regeneration: Oligodendrocytes and myelin as non-permissive substrates for neurite growth. Prog. Brain Res. 1988, 78, 363–370.
- Loschinger, J.; Bandtlow, C.E.; Jung, J.; Klostermann, S.; Schwab, M.E.; Bonhoeffer, F.; Kater, S.B. Retinal axon growth cone responses to different environmental cues are mediated by different second-messenger systems. J. Neurobiol. 1997, 33, 825–834.
- Bandtlow, C.E.; Schmidt, M.F.; Hassinger, T.D.; Schwab, M.E.; Kater, S.B. Role of intracellular calcium in NI-35-evoked collapse of neuronal growth cones. Science 1993, 259, 80–83.
- Habib, A.A.; Marton, L.S.; Allwardt, B.; Gulcher, J.R.; Mikol, D.D.; Hognason, T.; Chattopadhyay, N.; Stefansson, K. Expression of the oligodendrocyte-myelin glycoprotein by neurons in the mouse central nervous system. J. Neurochem. 1998, 70, 1704–1711.
- Kottis, V.; Thibault, P.; Mikol, D.; Xiao, Z.C.; Zhang, R.; Dergham, P.; Braun, P.E. Oligodendrocyte-myelin glycoprotein (OMgp) is an inhibitor of neurite outgrowth. J. Neurochem. 2002, 82, 1566–1569.
- Huang, J.K.; Phillips, G.R.; Roth, A.D.; Pedraza, L.; Shan, W.; Belkaid, W.; Mi, S.; Fex-Svenningsen, A.; Florens, L.; Yates, J.R., 3rd; et al. Glial membranes at the node of Ranvier prevent neurite outgrowth. Science 2005, 310, 1813–1817.
- Fernandez-Suarez, D.; Krapacher, F.A.; Andersson, A.; Ibanez, C.F.; Kisiswa, L. MAG induces apoptosis in cerebellar granule neurons through p75(NTR) demarcating granule layer/white matter boundary. Cell Death Dis. 2019, 10, 732.
- Shibata, A.; Wright, M.V.; David, S.; McKerracher, L.; Braun, P.E.; Kater, S.B. Unique responses of differentiating neuronal growth cones to inhibitory cues presented by oligodendrocytes. J. Cell Biol. 1998, 142, 191–202.
- McKerracher, L.; David, S.; Jackson, D.L.; Kottis, V.; Dunn, R.J.; Braun, P.E. Identification of myelin-associated glycoprotein as a major myelin-derived inhibitor of neurite growth. Neuron 1994, 13, 805–811.
- Tang, S.; Qiu, J.; Nikulina, E.; Filbin, M.T. Soluble myelin-associated glycoprotein released from damaged white matter inhibits axonal regeneration. Mol. Cell. Neurosci. 2001, 18, 259–269.
- Tang, S.; Woodhall, R.W.; Shen, Y.J.; deBellard, M.E.; Saffell, J.L.; Doherty, P.; Walsh, F.S.; Filbin, M.T. Soluble myelin-associated glycoprotein (MAG) found in vivo inhibits axonal regeneration. Mol. Cell. Neurosci. 1997, 9, 333–346.
- Mueller-Buehl, C.; Reinhard, J.; Roll, L.; Bader, V.; Winklhofer, K.F.; Faissner, A. Brevican, Neurocan, Tenascin-C, and Tenascin-R Act as Important Regulators of the Interplay Between Perineuronal Nets, Synaptic Integrity, Inhibitory Interneurons, and Otx2. Front. Cell Dev. Biol. 2022, 10, 886527.
- de Vivo, L.; Landi, S.; Panniello, M.; Baroncelli, L.; Chierzi, S.; Mariotti, L.; Spolidoro, M.; Pizzorusso, T.; Maffei, L.; Ratto, G.M. Extracellular matrix inhibits structural and functional plasticity of dendritic spines in the adult visual cortex. Nat. Commun. 2013, 4, 1484.
- Deepa, S.S.; Umehara, Y.; Higashiyama, S.; Itoh, N.; Sugahara, K. Specific molecular interactions of oversulfated chondroitin sulfate E with various heparin-binding growth factors. Implications as a physiological binding partner in the brain and other tissues. J. Biol. Chem. 2002, 277, 43707–43716.
- Frischknecht, R.; Heine, M.; Perrais, D.; Seidenbecher, C.I.; Choquet, D.; Gundelfinger, E.D. Brain extracellular matrix affects AMPA receptor lateral mobility and short-term synaptic plasticity. Nat. Neurosci. 2009, 12, 897–904.
- Pan, D.; Li, Y.; Yang, F.; Lv, Z.; Zhu, S.; Shao, Y.; Huang, Y.; Ning, G.; Feng, S. Increasing toll-like receptor 2 on astrocytes induced by Schwann cell-derived exosomes promotes recovery by inhibiting CSPGs deposition after spinal cord injury. J. Neuroinflamm. 2021, 18, 172.
- Asher, R.A.; Morgenstern, D.A.; Shearer, M.C.; Adcock, K.H.; Pesheva, P.; Fawcett, J.W. Versican is upregulated in CNS injury and is a product of oligodendrocyte lineage cells. J. Neurosci. 2002, 22, 2225–2236.
- Uhlin-Hansen, L.; Wik, T.; Kjellen, L.; Berg, E.; Forsdahl, F.; Kolset, S.O. Proteoglycan metabolism in normal and inflammatory human macrophages. Blood 1993, 82, 2880–2889.
- Jones, F.S.; Jones, P.L. The tenascin family of ECM glycoproteins: Structure, function, and regulation during embryonic development and tissue remodeling. Dev. Dyn. 2000, 218, 235–259.
- Monnier, P.P.; Sierra, A.; Schwab, J.M.; Henke-Fahle, S.; Mueller, B.K. The Rho/ROCK pathway mediates neurite growth-inhibitory activity associated with the chondroitin sulfate proteoglycans of the CNS glial scar. Mol. Cell. Neurosci. 2003, 22, 319–330.
- Shen, Y.; Tenney, A.P.; Busch, S.A.; Horn, K.P.; Cuascut, F.X.; Liu, K.; He, Z.; Silver, J.; Flanagan, J.G. PTPsigma is a receptor for chondroitin sulfate proteoglycan, an inhibitor of neural regeneration. Science 2009, 326, 592–596.
- Kimura-Kuroda, J.; Teng, X.; Komuta, Y.; Yoshioka, N.; Sango, K.; Kawamura, K.; Raisman, G.; Kawano, H. An in vitro model of the inhibition of axon growth in the lesion scar formed after central nervous system injury. Mol. Cell. Neurosci. 2010, 43, 177–187.
- Logan, A.; Berry, M.; Gonzalez, A.M.; Frautschy, S.A.; Sporn, M.B.; Baird, A. Effects of transforming growth factor beta 1 on scar production in the injured central nervous system of the rat. Eur. J. Neurosci. 1994, 6, 355–363.
- East, E.; Golding, J.P.; Phillips, J.B. A versatile 3D culture model facilitates monitoring of astrocytes undergoing reactive gliosis. J. Tissue Eng. Regen. Med. 2009, 3, 634–646.
- Renault-Mihara, F.; Mukaino, M.; Shinozaki, M.; Kumamaru, H.; Kawase, S.; Baudoux, M.; Ishibashi, T.; Kawabata, S.; Nishiyama, Y.; Sugai, K.; et al. Regulation of RhoA by STAT3 coordinates glial scar formation. J. Cell Biol. 2017, 216, 2533–2550.
- Bott, K.; Upton, Z.; Schrobback, K.; Ehrbar, M.; Hubbell, J.A.; Lutolf, M.P.; Rizzi, S.C. The effect of matrix characteristics on fibroblast proliferation in 3D gels. Biomaterials 2010, 31, 8454–8464.
- Tran, A.P.; Warren, P.M.; Silver, J. The Biology of Regeneration Failure and Success After Spinal Cord Injury. Physiol. Rev. 2018, 98, 881–917.
- Li, X.; Yang, B.; Xiao, Z.; Zhao, Y.; Han, S.; Yin, Y.; Chen, B.; Dai, J. Comparison of subacute and chronic scar tissues after complete spinal cord transection. Exp. Neurol. 2018, 306, 132–137.
- Weidner, N.; Grill, R.J.; Tuszynski, M.H. Elimination of basal lamina and the collagen "scar" after spinal cord injury fails to augment corticospinal tract regeneration. Exp. Neurol. 1999, 160, 40–50.
- Gonzalez-Perez, F.; Udina, E.; Navarro, X. Extracellular matrix components in peripheral nerve regeneration. Int. Rev. Neurobiol. 2013, 108, 257–275.
- Anderson, M.A.; Burda, J.E.; Ren, Y.; Ao, Y.; O’Shea, T.M.; Kawaguchi, R.; Coppola, G.; Khakh, B.S.; Deming, T.J.; Sofroniew, M.V. Astrocyte scar formation aids central nervous system axon regeneration. Nature 2016, 532, 195–200.
- Faulkner, J.R.; Herrmann, J.E.; Woo, M.J.; Tansey, K.E.; Doan, N.B.; Sofroniew, M.V. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J. Neurosci. 2004, 24, 2143–2155.
- Hemati-Gourabi, M.; Cao, T.; Romprey, M.K.; Chen, M. Capacity of astrocytes to promote axon growth in the injured mammalian central nervous system. Front. Neurosci. 2022, 16, 955598.
- Bloom, O. Non-mammalian model systems for studying neuro-immune interactions after spinal cord injury. Exp. Neurol. 2014, 258, 130–140.
- Zukor, K.A.; Kent, D.T.; Odelberg, S.J. Meningeal cells and glia establish a permissive environment for axon regeneration after spinal cord injury in newts. Neural. Dev. 2011, 6, 1.
- Hara, M.; Kobayakawa, K.; Ohkawa, Y.; Kumamaru, H.; Yokota, K.; Saito, T.; Kijima, K.; Yoshizaki, S.; Harimaya, K.; Nakashima, Y.; et al. Interaction of reactive astrocytes with type I collagen induces astrocytic scar formation through the integrin-N-cadherin pathway after spinal cord injury. Nat. Med. 2017, 23, 818–828.
- Rowland, J.W.; Hawryluk, G.W.; Kwon, B.; Fehlings, M.G. Current status of acute spinal cord injury pathophysiology and emerging therapies: Promise on the horizon. Neurosurg. Focus 2008, 25, E2.
- Ahuja, C.S.; Wilson, J.R.; Nori, S.; Kotter, M.R.N.; Druschel, C.; Curt, A.; Fehlings, M.G. Traumatic spinal cord injury. Nat. Rev. Dis. Primers 2017, 3, 17018.
- Ehlers, M.D. Deconstructing the axon: Wallerian degeneration and the ubiquitin-proteasome system. Trends Neurosci. 2004, 27, 3–6.
- Sadek, A.R.; Nader-Sepahi, A. Spinal Arachnoid Cysts: Presentation, management and pathophysiology. Clin. Neurol. Neurosurg. 2019, 180, 87–96.
- Kwiecien, J.M.; Dabrowski, W.; Yaron, J.R.; Zhang, L.; Delaney, K.H.; Lucas, A.R. The Role of Astrogliosis in Formation of the Syrinx in Spinal Cord Injury. Curr. Neuropharmacol. 2021, 19, 294–303.
- Scivoletto, G.; Masciullo, M.; Pichiorri, F.; Molinari, M. Silent post-traumatic syringomyelia and syringobulbia. Spinal Cord Ser. Cases 2020, 6, 15.
- Lu, Z.; Fu, L.; Fan, X. Spinal cord stimulation for treatment of neuropathic pain associated with syringomyelia. Asian J. Surg. 2022. Online ahead of print.
More
Information
Subjects:
Neurosciences
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
4.5K
Revisions:
2 times
(View History)
Update Date:
23 Dec 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No