Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Agata M. Sakowicz | -- | 4474 | 2022-12-22 14:51:42 | | | |
| 2 | Sirius Huang | -1 word(s) | 4473 | 2022-12-23 04:05:11 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Sakowicz, A.; Bralewska, M.; Kamola, P.; Pietrucha, T. Animal Models of Preeclampsia. Encyclopedia. Available online: https://encyclopedia.pub/entry/39103 (accessed on 25 June 2026).
Sakowicz A, Bralewska M, Kamola P, Pietrucha T. Animal Models of Preeclampsia. Encyclopedia. Available at: https://encyclopedia.pub/entry/39103. Accessed June 25, 2026.
Sakowicz, Agata, Michalina Bralewska, Piotr Kamola, Tadeusz Pietrucha. "Animal Models of Preeclampsia" Encyclopedia, https://encyclopedia.pub/entry/39103 (accessed June 25, 2026).
Sakowicz, A., Bralewska, M., Kamola, P., & Pietrucha, T. (2022, December 22). Animal Models of Preeclampsia. In Encyclopedia. https://encyclopedia.pub/entry/39103
Sakowicz, Agata, et al. "Animal Models of Preeclampsia." Encyclopedia. Web. 22 December, 2022.
Copy Citation
Preeclampsia is a disorder associated with pregnancy and entails a high risk of maternal and foetal mortality and morbidity. In vivo studies on the pathology of gestation, including preeclampsia, often use small mammals such as rabbits or rodents, i.e., mice, rats, hamsters, and guinea pigs. The key advantage of these animals is their short reproductive cycle; in addition, similar to humans, they also develop a haemochorial placenta and present a similar transformation of maternal spiral arteries.
mouse
placenta
preeclampsia
rabbit
rat
rodents
trophoblast
1. Introduction
The long period between the first event initiating the development of preeclampsia and the resulting clinical symptoms indicates that the development of preeclampsia is a multi-stage process that requires a constellation of events in humans. As such, it is difficult to find an animal model that reflects all of the stages of pathological human gestation; indeed, the period of gestation, the process of implantation, and placental formation as well as hormonal or immunological status vary considerably between animal species and humans. In this sense, all known animal models are therefore incomplete, and even non-human primate animal models presenting spontaneous preeclampsia have some limitations [1]. This restricts the utility of animal models to the investigation of selective fragments of the pathomechanism of preeclampsia [2].
As presented in Table 1, the most commonly used rodents for the study of preeclampsia are mice and rats. Hamsters and guinea pigs are not popular for studies of pregnancy disorders despite demonstrating some common features with human pregnancy, i.e., cytotrophoblast plugs (hamster) or a hemimonochorial placenta (guinea pig). This might be related to the difficulties in the process of measuring blood pressure for these animals. Rats and mice have a long tails, allowing for the non-invasive measurement of blood pressure. Invasive methods, such as telemetric probes, entail surgery with a high risk of mortality due to thrombosis, blood loss, or systemic infection. Moreover, these methods increase the stress experienced by the animal, and the size of the probe has to be matched to the size of the animal, which changes considerably over the course of gestation [3][4]. Additionally, the experimental tools are not readily available for mechanistic analyses of placentation in hamsters and guinea pigs [5], and the downstream laboratory examinations of samples obtained from these animals require reagents dedicated to the studied animals. A number of reagents, e.g., the antibodies used for Western blot or ELISA techniques, do not allow for the examination of guinea pig proteins, requiring the creation and purchase of special reagents.
Table 1. The generation of preeclampsia by the induction of various aspects of the disease in animal models.
| Species | Inducing Factor | Time of PE Induction | HA | P or A/C Ratio |
Ref |
|---|---|---|---|---|---|
| Incorrect trophoblast invasion | |||||
| Rat | 0.5 µg/kg LPS | 5 GD | + | + | [6][7] |
| Rat | Transgenic rats hAngiotensinogen female x hRenin male | spontaneously | + | + | [8][9] |
| Rat | Stroke-prone spontaneously hypertensive rat (SHRSP) | spontaneously | + | + | [10] |
| Mouse | LPS 20 µg/kg | 7.5–17.5 GD | + | + | [11] |
| Mouse | BPH5 mouse | spontaneously | + | + | [12] |
| Guinea pig | Exposure to hypoxia 10.5% O2 | 28–30 GD up to term | + | ns | [13] |
| Placental ischaemia | |||||
| Rat | RUPP model | 14 GD | + | -, ns | [14][15] |
| Rat | Selective RUPP model | 14 GD | + | ns | [16] |
| Mouse | RUPP model | 14.5 GD | + | + | [17] |
| Rabbit | RUPP model | 25 GD | + | - | [18] |
| Incorrect angiogenesis | |||||
| Rat | Injection—adenovirus expressing sFlt-1 | 8 or 9 GD | + | + | [19] |
| Rat | Injection—adenovirus sFtl-1 and/or sEng | 8 or 9 GD | + | + | [20] |
| Rat | Injection—Suramin (an inhibitor of angiogenesis) | 10 and 11 GD | + | - | [21] |
| Mouse | Injection—adenovirus expressing stabilised HIF-1α | 8 GD | + | + | [22] |
| Mouse | Injection—exosomes of preeclamptic women | 5.5; 10.5; 15.5 GD | + | + | [23] |
| Mouse | Injection—adenovirus carrying sFlt-1 | 4; 8; 9 GD | + | + | [24] |
| Mouse | Injection—adenovirus carrying sFlt-1 | 8 GD | + | + | [25] |
| Mouse | Injection into blastocyst lentivirus carrying human sFlt-1 | in vitro | + | + | [26] |
| Inflammation | |||||
| Rat | TNFα infusion | 14–19 GD | + | ns | [27] |
| Rat | TNFα infusion | 14 GD | + | - | [28] |
| Rat | IL6 infusion | 14–19 GD | + | - | [29] |
| Mouse | Injection of purified IgG from PE women | 13 and 14 GD | + | + | [30] |
| Mouse | TNFα injection | 13 GD | + | + | [31] |
| Mouse | IL10 deficient mouse | spontaneously | + | + | [32] |
| Mouse | BPH5 mouse | spontaneously | + | + | [33] |
| Endothelial dysfunction | |||||
| Rat | L-NAME in drinking water | 8–19 GD | + | + | [34] |
| Rat | L-NAME injection | 9-20 GD or 10-20 GD | + | + | [35] |
| Rat | AT1-AA antibody injection | 13 and 14 GD | + | + | [36] |
| Mouse | L-NAME injection | 7–18 GD | + | + | [37] |
| Mouse | AT1-AA antibody injection | 13 GD | + | + | [38] |
| Mouse | eNOS knockout mice | spontaneously | + | + | [39] |
PE—preeclampsia; GD—gestational day; HA—hypertension; P or A/C—proteinuria or albumin to creatinine ratio in urine; LPS—lipopolysaccharide; RUPP—reduced uterine perfusion; sFlt-1—soluble fms-like tyrosine kinase 1; sEng—soluble endoglin-1; TNFα—tumour necrosis factor alpha; IL6—interleukin 6; IL10—interleukin 10; L-NAME—N-nitro-L-arginine methyl ester, an inhibitor of NO synthase (NOS); AT1-AAs antibodies—antibodies against the angiotensin II receptor type 1a (AT1 receptor) localised on endothelial cells; IgG—immunoglobin G; BPH5—high blood pressure (BPH)/5 mouse; eNOS—endothelial nitric oxide synthase. +—present, -—not measured, ns—non significant differences between studied groups.
2. An Incorrect Trophoblast Invasion Model of Preeclampsia
Although the main source of preeclampsia development is believed to be incorrect trophoblast invasion, it is not currently clear whether this process is the primary defect or a consequence of other processes related to the decidualisation or preparation of maternal vessels for contact with cytotrophoblast [40]. Additionally, no single factor in the maternal blood or uterus has been identified that inhibits cytotrophoblast invasion and maternal spiral artery transformation. Therefore, any insights regarding shallow trophoblast invasion and the incorrect remodelling of uterine vessels obtained after removing the placenta cannot be predicted at the beginning of the study, and they should therefore be regarded merely as side effects of preeclampsia induction in laboratory animals.
The most common preeclamptic animal models presenting insufficient trophoblast invasion are related to the generation of inflammation at the beginning or middle period of pregnancy (Table 1). In these animal models, the inflammatory state is generally generated by a low dose of lipopolysaccharides (LPS) obtained from Gram-negative bacteria, i.e., Escherichia coli. In rat models, hypertension is obtained by the intravenous injection of a single dose of LPS (0.5 µg/kg body mass) on the fifth gestational day (GD) [6] or of 1 µg/kg body mass at day 14 [41] or is generated by a single intraperitoneal infusion of a low-dose of LPS (10 µg/kg body mass) at GD 13.5 followed by daily injections of higher doses (40 µg/kg body mass) until GD 16.5 [42]. Similarly, in mouse models, trophoblast invasion impairment and spiral artery remodelling can be induced by daily intraperitoneal LPS treatment (20 µg/kg body mass) from 7.5 to 17.5 [11]; this also generates a preeclampsia-like phenotype.
High blood pressure is typically observed a few hours after the first LPS injection [11][41][42]. Interestingly, hypertension is only observed in the population of pregnant animals; the non-pregnant animals do not respond to the supplementation of LPS with an elevation in blood pressure. It is possible that the inflammation generated by LPS acts as the first signal initiating the pathomechanism of preeclampsia, which is followed by incorrect decidualisation or trophoblast invasion. The impairment of decidualisation and the transformation of maternal uterine vessels may be responses by the foetus/mother to the ongoing inflammatory process in the maternal environment [43]. It is believed that chronic, often asymptomatic, the infection might play a significant role in the development of preeclampsia. Therefore, the association between periodontal disease and the outcome of preeclampsia is not surprising. Chronic oral infection predisposes the patient to systematic illnesses, especially those associated with the cardiovascular system; one example is atherosclerosis, which also activates maternal inflammatory mechanisms [44][45]. These mechanisms are also activated by other chronic disorders, including obesity, diabetes, hypertension prior to pregnancy, or autoimmunological diseases, which are known risk factors for the development of preeclampsia in humans.
Inflammation, especially that located in the uterine tissue, might be intensified by the hypoxic conditions dominating in the uterus at the beginning of all pregnancies. This temporally limited hypoxic state, i.e., about 2% O2, is necessary for the correct process of trophoblast invasion and proliferation and for the remodelling of the maternal vessels. However, chronic hypoxia seems to be a negative regulator of maternal vessel transformation. Guinea pig mothers maintained in hypoxic conditions, i.e., 10.5% O2 from 20 GD until the middle of gestation, demonstrated elevated trophoblastic cell proliferation, but with impaired invasive potential and maternal vessel remodelling [13]. Similar results were observed for populations of pregnant rats maintained in hypoxic conditions, i.e., 10.5% O2 between days 6 and 21 of gestation. These rats presented high blood pressure, which increased significantly on day 12 of pregnancy and demonstrated a higher protein-to-creatinine ratio in the urine compared to controls. The animals presented the impairment of trophoblast invasion or uteroplacental vessel remodelling and also demonstrated foetal growth restriction [46]. In addition, chronic low-oxygen tension was found to significantly impact the elevation of endothelin-1 level in maternal plasma, one of the strongest vasoconstriction factors secreted both by ischaemic placenta and maternal endothelial cells [46].
The hypothesis that incorrect trophoblast invasion might induce preeclampsia development has also been verified in spontaneously induced preeclampsia animal models, including those based on stroke-prone spontaneously hypertensive rats (SHRSP). These rats demonstrated hypertension before pregnancy and a significant elevation in blood pressure compared to baseline at day 13 of gestation. They also manifested defects in trophoblast invasion and uteroplacental vessel transformation, as indicated by histochemical studies of placentas [10].
Similar to SHRSP rats, BPH/5 mice, i.e., a genetically pre-hypertensive animal model obtained by mating the brother and sister of hypertensive BPH/2 mice, also demonstrate problems with trophoblast invasion and spiral artery remodelling [12]. In addition to the placentation problem, BPH/5 mice also exhibit excessive inflammation in the implantation site, resulting in litters with a low number of pups with low weight [33].
Interestingly, the histochemical results obtained from SHRSP rats and BPH/5 mice are contradicted by another Sprague Dawley rat model derived by mating animals harboring one human renin (hRen) gene or human angiotensinogen gene (hAogen), i.e., genes coding for proteins regulating blood pressure in humans. After mating with a male (hRen), the dams (hAogen) developed hypertension at day 13 of gestation and presented albuminuria; in addition, similar to preeclamptic women, the clinical symptoms of preeclampsia disappeared after gestation [8][9]. The dams also delivered smaller litters with lower mean weight compared to the controls created by mating a normal female rat with a male rat harboring hRen or with a normal male rat. Interestingly, rats presenting the clinical features of preeclampsia demonstrated significantly deeper trophoblast invasion than the controls, determined at GD 18, i.e., on the day of maximal invasion of trophoblasts into the mesometrial triangle. These preeclamptic dams also presented a high rate of fibrinoid deposition and lower numbers of endothelial maternal cells in the uterine spiral arteries; both features are recognised as desirable physiological changes associated with vascular remodelling in both human and rat pregnancies [8].
These findings suggest that the pathomechanism of preeclampsia varies slightly between cases depending on the first signal of activation [47][48][49][50]. It is possible that defects in the regulation of the renin–angiotensin–aldosterone (RAA) system may increase the risk of preeclampsia but via different mechanisms depending on the nature of the defects. In pregnancy, the RAA system not only regulates maternal blood pressure and fluid homeostasis, but it is also believed to play a role in processes associated with the development and functioning of the placenta, such as in angiogenesis, cytotrophoblast proliferation, maternal spiral artery remodelling, placental nutrient transport, and placental hormone secretion [51][52]. Therefore, individual defects in the RAA system could influence the development of either early-onset or late-onset preeclampsia [53]; this could be one of the reasons why defects in trophoblast invasion are not always as evident for late-onset preeclampsia as in cases developing before week 34 of gestation
3. The Placental Ischaemia in Animal Models for Preeclampsia
Although previous models indicate an association between disturbances in trophoblast invasion, maternal spiral remodelling, and preeclampsia development, they do not constitute sufficient proof whether improper placentation, which can lead to chronic placental ischaemia, can fully account for the clinical symptoms of preeclampsia. However, this can be confirmed by animal models based on the mechanical non-complete occlusion of vessels delivering maternal blood to the uteroplacental unit. This reduced uterine perfusion pressure (RUPP) model is characterised by an approximately 40% reduction in uterine perfusion. Depending on the study, vessel occlusion was generated by silver clips or ligation. The three most common locations (Figure 1) were around the aorta right above the iliac bifurcation and around the left and right uterine arcade at the ovarian artery before the first segmental artery to prevent the adaptive increases in blood flow in the ovarian arteries (the conventional RUPP model, Figure 1b) around the uterine arterial and venous branches of the vascular arcade at their ovarian vessels or uterine vessels (the new RUPP model, Figure 1c) or on the ovarian and uterine arteries (the selective RUPP model) (Figure 1d). Additional RUPP model variants are given elsewhere [54]. Controls for all the RUPP animals were created by sham-operated animals or were non-pregnant females that underwent RUPP surgery.
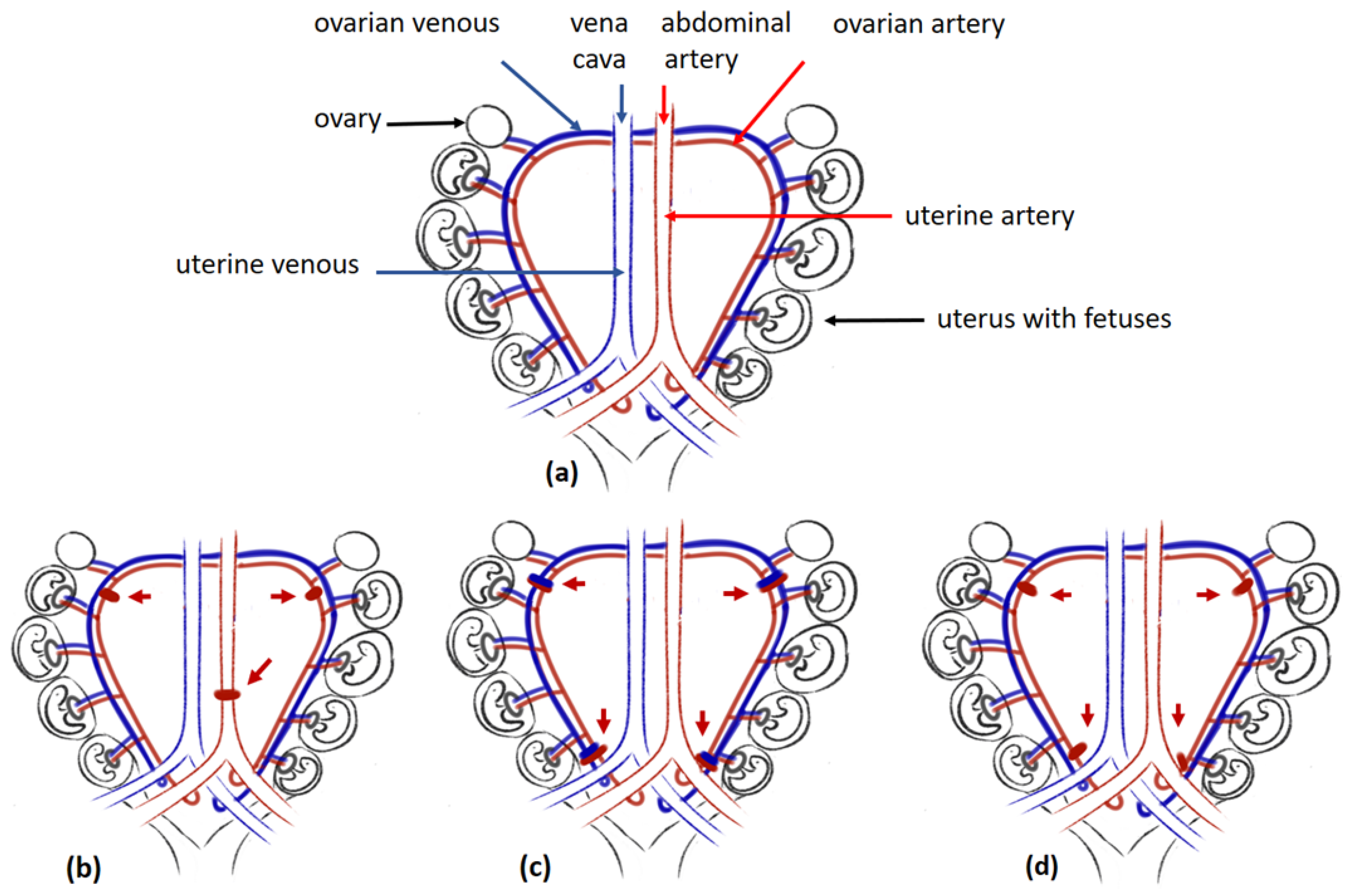
Figure 1. The RUPP models: (a) Uterus of rat, (b) conventional RUPP model, (c) new RUPP model, (d) selective RUPP model. Legend: red arrow pointing at silver clips placed around vessels; red clips are placed around the arteries; blue clips are placed around veins.
Although the method of occlusion of maternal vessels varies between studies, in most cases, surgery was performed about seven to nine days before the delivery date typical of the species, i.e., about 11.5–14.5 days postcoitum for mice, at about gestational day 14 for rats, or at about day 22 for rabbits [14][16][17][18][55]. Interestingly, while both mouse and rabbit models presented elevated blood pressure 24 h after surgery, hypertension took three or more days to develop in rats, and it was maintained until the day of termination of gestation [16][18][56]. Although RUPP animals develop other clinical features indicative of preeclampsia, e.g., proteinuria, glomerular defects, or foetal growth restriction [17][54][56][57], the precise nature of the changes varies between RUPP variants. For example, among mice treated with a new RUPP model (Figure 1c) in which only some places of ligation were preserved (1) around the ovarian vessels distal to the branches to the ovaries (O,O mouse model) or (2) around the uterine vessels (U,U mouse model), only the latter presented characteristic changes in sFlt1 and PlGF placental gene expression, as observed in preeclamptic women [17].
Similarities can be found between animal models based on conventional RUPP surgery and preeclamptic women with regard to the profile of factors regulating angiogenesis, placental development, and endothelial dysfunction as well as an immunological maternal reaction. This profile includes elevated levels of soluble Flt-1, one of the most important anti-angiogenetic placental-derived factors [14][54][55][58]. Moreover, similar to human preeclamptic pregnancies, ischaemic placentas are known to downregulate PlGF expression; both PIGF and VEGF were found depleted in RUPP rat serum [54][58].
RUPP rats demonstrate a similar serum inflammatory cytokine profile to that of human preeclamptic mothers, with higher levels of IL-6 and TNFα; however, this is not noted in RUPP mice. These cytokines stimulate the production of autoantibodies against the angiotensin II-type receptor (AT1-AA), whose level is also elevated in preeclamptic pregnancies [59][60][61][62].
Although RUPP animals share a number of common features with human preeclamptic mothers, this model is not ideal. A key disadvantage is that as the RUPP model is created by the mechanical occlusion of maternal vessels, it is not suitable for the investigation of the patomechanism of preeclampsia placed before the day of surgery, i.e., generally before days 11.5–14 of gestation in rodents and before day 22 in rabbits. Secondly, some studies indicate that the occlusion of uterine maternal vessels not only inhibits blood flow in the uterus but also in other organs, i.e., heart, kidney, or brain [54]. Finally, this model may not be applicable for testing new drugs whose mechanism of action might be related to increased blood flow to the placenta. However, despite this, the RUPP model delivers unquestionable proof that the ischaemic placenta secretes a number of factors regulating the process of angiogenesis, whose incorrect levels in maternal blood are linked with the pathomechanism of preeclampsia.
4. Models of Preeclampsia Employing the Incorrect Angiogenesis
RUPP models indicate that the ischaemic placenta produces elevated levels of anti-angiogenic factors, such as sFlt-1 and soluble endoglin 1 (sEng1), and that their levels negatively correlate with those of PlGF and VEGF in maternal circulation [63]. The roles of these anti-angiogenic factors in preeclampsia have been determined by stimulating animals with these agents (Table 1).
The animal models mimicking placental disorders are generated by the infusion of sFlt1 or sFlt1 and sEng1 particles. Intraperitoneal injections of recombinant sFlt1 between days 13 and 18 of gestation were found to generate hypertension in pregnant rats; in addition, treatment with an adenovirus carrying sFlt1 stimulated hypertension in a mouse population at day 8 of gestation [25][64][65]. The stimulated animals also presented features of glomerular damage, with 70% amelioration in the glomerular production of nitrate oxide being observed in some models [24][66]. Interestingly, sFlt1 influences the generation of high blood pressure in both pregnant and non-pregnant animals: non-pregnant Sprague Dawley rats and Balb/c mice treated by adenoviruses carrying the sFlt1 were both found to demonstrate a preeclamptic phenotype [19][24]. These animal models confirm that placentally derived particles such as sFlt1 or sEng play a significant role in the regulation of blood pressure. Moreover, these models are suitable for the investigation of new therapies based on targeting sFlt1 to reduce pregnancy-induced hypertension [153[67][68].
Correct cell–cell communication is essential for ensuring the proper functioning of all organisms. Exosomes, extracellular vehicles, are secreted by most cells to transport various substances, e.g., mRNA, miRNA, DNA, or proteins, for short or long distances [69]. The exosomes secreted into maternal blood by the preeclamptic placenta are sources of anti-angiogenic factors such as sFlt1 and sEng1. Research on C57/BL6 pregnant mice indicates that the exosomes isolated from the serum of preeclamptic women (PE-exosomes) can stimulate hypertension and renal disorders in pregnant animals. These mice also presented lower litter weights, probably as a consequence of the extensive vascular damage caused to the vessels localised within the labyrinth of the placenta [23]. Both sFlt1 and sEng1 were also found to induce severe preeclampsia in Sprague Dawley rats [20]. A significant body of evidence indicates that sFlt1 neutralises proangiogenic factors circulating in maternal blood, such as the free forms of VEGF and PlGF; in addition, sEng1 not only negatively regulates angiogenesis but also increases the permeability of the maternal vessels and blocks the activation of endothelial nitric oxide synthase (eNOS), downregulating the production of nitric oxide, one of the strongest vasodilators [20].
The inhibition of angiogenesis was found to influence the development of preeclampsia in a group of pregnant Sprague Dawley rats treated by Suramin, an angiogenesis inhibitor antagonizing VEGF, platelet-derived growth factor, or fibroblast growth factor [21]. The rats demonstrated hypertension accompanied by placental dysfunction, renal failure, and reductions in foetal body mass. Interestingly, the Suramin-treated rats demonstrated over 50% lower sFlt1 levels compared to the controls. This may be the reason for the smaller litter numbers carried by the treated rats and thus the smaller number of placentas secreting sFlt1 compared to the controls [21]. These findings might also suggest that other unidentified maternal factors appearing in maternal blood might make preeclampsia development more likely. Indeed, although preeclamptic women demonstrate the classical pattern of high levels of circulating sFlt1 and low levels of free PlGF, some studies suggest that preeclampsia is also associated with low sFlt1 and high PlGF levels [70]. It is possible that the placental cells of these women secrete other factors that deplete VEGF levels and thus disturb angiogenesis. Treatment with sFlt1 inhibitors will certainly be ineffective in such populations; therefore, models based on Suramin treatment might be a promising basis for designing personal therapies for these rare groups of preeclamptic women.
5. Inflammatory Factors Exert Preeclampsia in Animal Models
Preeclampsia is clearly related to the activation of immunological systems and is manifested by increases in TNFα, IL6, or IL1 levels in maternal circulation. Such inflammation accompanies preeclampsia from the beginning of gestation and continues throughout the course of preeclamptic pregnancy. A number of studies indicate that maternal pre-pregnancy disorders associated with inflammation, such as obesity, diabetes, or autoimmunological diseases, predispose one to the development of preeclampsia. Early inflammation has been confirmed to induce hypertension in animal models treated by LPS at the beginning of gestation (Table 1). The treated rats and mice developed hypertension and trophoblast invasion disorders, as discussed in the previous section. “An incorrect trophoblast invasion model of preeclampsia”.
It seems that inflammation does not only influence the development of preeclampsia, but the mechanism of its action depends the strength and the time of its initiation. A significant body of evidence indicates that high levels of sFlt1 not only result from placental ischaemia but also from the response of placental cells to inflammation. Sprague Dawley rats administered 50 ng/day of TNFα for five days beginning from day 14 of gestation presented a significant increase in the sFlt1 level in maternal blood and higher mean arterial pressure (MAP) in comparison to not-treated controls. This elevated sFlt1 level, caused by chronic infusion of TNFα, was reduced after maternal injections of a soluble TNFα receptor [71].
Interestingly, the infusion of a single dose of TNFα after the middle of gestation, i.e., at day 13.5 of gestation-induced placental hypoxia in C57BL/6JArc mice, resulted in the elevated expression of hypoxia inducible factor 1 (HIF1) in all sections of the mouse placentas, i.e., the labyrinth, junctional zone, and decidua. Although significant expression of the gene coding for Flt1 was noticed in ischaemic placentas, surprisingly, the levels of Flt1 in dam serum fell, albeit insignificantly [31].
The injection of inflammatory factors into pregnant animals not only influences the placenta but also forces the maternal organism to generate agents similar to those observed in preeclamptic women. Daily infusions of TNFα to rats between days 14 and 19gestation induced the significant generation of AT1-AA antibodies, which are responsible for the activation of the angiotensin II type I (AT1R) receptor, which is found during vasoconstriction, salt-water retention, and aldosterone secretion [62][72]. Elevated AT1-AA antibody levels are also typical of human pregnancies complicated by preeclampsia or abnormal uterine perfusion. Interestingly, the chronic infusion of TNFα into non-pregnant rats did not increase the production of AT1-AA antibodies, suggesting that the elevation in AT1-AA occurring in maternal serum in response to inflammation is restricted to pregnancy [62]. Similarly, a relationship between pregnancy-induced hypertension and high AT1-AA levels was observed in Sprague Dawley rats after IL6 infusion, suggesting that the production of these antibodies depends on the presence of unwanted inflammation in pregnancies [29].
The reason why AT1-AA generation is augmented in pregnancies complicated by inflammation remains a mystery. Nevertheless, it is believed that these antibodies and various inflammatory factors have a direct impact on the dysregulation of the maternal endothelium. AT1-AA antibodies mainly support the process of vasoconstriction by stimulating endothelial cells to produce endothelin-1 [73]. Similar effects, such as the elevated expression of preproendothelin mRNA in the kidney, placenta, and aorta, were obtained by daily the infusion of TNFα for five days beginning from day 14 of gestation [74]. Moreover, TNFα is perceived as a mediator in the vascular contraction influencing endothelial dysfunction. It inhibits acetylcholine- and bradykinin-induced vascular relaxation as well as nitrate production in endothelial cells, especially in pregnant rats [75]. Although animal models based on the supplementation of inflammatory factors indicate that both chronic and acute inflammation play significant roles in the development of the clinical features of preeclampsia, these models are not good candidates for determining whether endothelial dysfunction, a consequence of inflammation, is sufficient to generate the clinical symptoms of the disease.
6. Endothelial Dysfunction: The Last Step in the Generation of Preeclampsia
A significant body of evidence indicates that endothelial dysfunction has a key influence on the development of the preeclamptic phenotype. It is known that the vasodilatory propriety of endothelial cells influences the regulation of blood pressure, especially during pregnancy, which is characterised by high cardiac output, blood volume, and heart rate compared to a pre-pregnancy baseline but normal or slightly lower blood pressure. The most important regulator of blood pressure is nitric oxide (NO), which is produced in endothelial cells from L-arginine by enzyme endothelial nitric oxide synthase (eNOS). The eNOS knockout mice present the preeclamptic phenotype; however, hypertension is recognised both before and during pregnancy [39][50][76], and proteinuria develops with late gestation, i.e., at 18.5 GD [39]. Although the development of hypertension induced by pregnancy is faint, these mice present other clinical features typical of preeclampsia. The most commonly reported findings are foetal growth restriction and reduction in uterine blood flow. Moreover, hypoxia and reduced nutrient transport capacity are characteristic conditions observed in the placentas of mice lacking genes coding for endothelial nitric oxide synthase [39][77]. The production of NO could be blocked by L-arginine analogues such as nitro-L-arginine methyl ester (L-NAME), which causes NO deficiency by inhibiting NOS activity [34]. The reaction of animal models depends on the mode of L-NAME supplementation: one study on a rat model found that L-NAME treatment at the beginning of gestation, i.e., between days 9 and 11, resulted in a lower rise in mean blood pressure (MAP) compared to later treatment between days 18 and 20 [78]. This suggests that the endothelium has a significant impact on the regulation of blood pressure, especially in the last trimester of pregnancy. This period of gestation is characterised by high blood volume in maternal circulation; as such, the regulation of vascular tone might play a significant role in maintaining correct blood pressure. Numerous studies indicate that supplementation with L-NAME induces hypertension and proteinuria in pregnant rodents and rabbits [34][79][80][81][82], and some animals present additional symptoms, i.e., thrombocytopenia and intrauterine growth restriction, which are indicative of preeclampsia in humans [34]. Moreover, rats treated with L-NAME demonstrate a higher sFlt1 to PlGF ratio in maternal serum than untreated controls [35]; however, this ratio was insignificantly elevated for mice treated with L-NAME [83].
The offspring born from mothers treated with L-NAME during pregnancy were found to suffer from hypotension for the first two weeks of their postnatal life [84]. A similar observation was noted in the first hours after delivery for human infants born from preeclamptic pregnancies [85]. Interestingly, gravid and non-pregnant rats demonstrated a similar response to L-NAME infusion [86]: although both groups demonstrate a preeclampsia-like phenotype, the presence of hypertension and proteinuria in virgins contradicts the main assumption of the diagnosis of preeclampsia, as it is reserved for the gestational period. This suggests that endothelial dysfunction is necessary to provoke the development of the clinical symptoms of preeclampsia. Unfortunately, L-NAME animal models are not suitable for providing a full picture of this disease, especially the aspects related with placental dysfunction. Indeed, the placentas obtained from a murine L-NAME model did not present any alternations in the labyrinth or junctional zones or in the maternal deciduae. The only changes were observed for sinuses, which were narrower than those of the control dams [83].
The mode of induction of hypertension for these models is also questionable. Although pregnancies not complicated by hypertension demonstrate an amelioration in NO production, the association between the downregulation of NO and preeclampsia remains ambiguous [61][87]. However, the L-NAME model is a good candidate model for investigating potential drugs that might be used to modulate the disturbed pathways in the endothelium of preeclamptic women [88]. Such drugs might be desirable for treating women manifesting the whole spectrum of preeclamptic symptoms; indeed, by modifying the disturbed pathways in the maternal endothelium, it may be possible to suppress disease-related symptoms and thus increase the length of gestation, giving the foetus more time to thrive.
References
- Kumasawa, K. Animal Models in Preeclampsia. In Preeclampsia Basic, Genomic, and Clinical; Saito, S., Ed.; Springer: Berlin/Heidelberg, Germany, 2018; pp. 141–155. ISBN 9789811058912.
- Bakrania, B.A.; George, E.M.; Granger, J.P. Animal models of preeclampsia: Investigating pathophysiology and therapeutic targets. Am. J. Obstet. Gynecol. 2022, 226, S973–S987.
- Bogdan, S.; Luca, V.; Ober, C.; Melega, I.; Pestean, C.; Codea, R.; Oana, L. Comparison among different methods for blood pressure monitoring in rats: Literature review. Bull. Univ. Agric. Sci. Vet. Med. Cluj-Napoca Vet. Med. 2019, 76, 5.
- Chau, K.; Welsh, M.; Makris, A.; Hennessy, A. Progress in preeclampsia: The contribution of animal models. J. Hum. Hypertens. 2022, 36, 705–710.
- Soares, M.J.; Chakraborty, D.; Rumi, M.A.K.; Konno, T.; Renaud, S.J. Rat placentation: An experimental model for investigating the hemochorial maternal-fetal interface. Placenta 2012, 33, 233–243.
- Gong, P.; Liu, M.; Hong, G.; Li, Y.; Xue, P.; Zheng, M.; Wu, M.; Shen, L.; Yang, M.; Diao, Z.; et al. Curcumin improves LPS-induced preeclampsia-like phenotype in rat by inhibiting the TLR4 signaling pathway. Placenta 2016, 41, 45–52.
- Xue, P.; Zheng, M.; Gong, P.; Lin, C.; Zhou, J.; Li, Y.; Shen, L.; Diao, Z.; Yan, G.; Sun, H.; et al. Single administration of ultra-low-dose lipopolysaccharide in rat early pregnancy induces TLR4 activation in the placenta contributing to preeclampsia. PLoS ONE 2015, 10, e0124001.
- Geusens, N.; Verlohren, S.; Luyten, C.; Taube, M.; Hering, L.; Vercruysse, L.; Hanssens, M.; Dudenhausen, J.W.; Dechend, R.; Pijnenborg, R. Endovascular Trophoblast Invasion, Spiral Artery Remodelling and Uteroplacental Haemodynamics in a Transgenic Rat Model of Pre-eclampsia. Placenta 2008, 29, 614–623.
- Dechend, R.; Gratze, P.; Wallukat, G.; Shagdarsuren, E.; Plehm, R.; Bräsen, J.H.; Fiebeler, A.; Schneider, W.; Caluwaerts, S.; Vercruysse, L.; et al. Agonistic autoantibodies to the AT1 receptor in a transgenic rat model of preeclampsia. Hypertension 2005, 45, 742–746.
- Barrientos, G.; Pussetto, M.; Rose, M.; Staff, A.C.; Blois, S.M.; Toblli, J.E. Defective trophoblast invasion underlies fetal growth restriction and preeclampsia-like symptoms in the stroke-prone spontaneously hypertensive rat. Mol. Hum. Reprod. 2017, 23, 509–519.
- Li, G.; Ma, L.; Lin, L.; Wang, Y.L.; Yang, H. The intervention effect of aspirin on a lipopolysaccharide-induced preeclampsia-like mouse model by inhibiting the nuclear factor-κB pathway. Biol. Reprod. 2018, 99, 422–432.
- Sones, J.L.; Yarborough, C.C.; Besso, V.O.; Lemenze, A.; Douglas, N.C. Genotypic analysis of the female BPH/5 mouse, a model of superimposed preeclampsia. PLoS ONE 2021, 16, e0253453.
- Thompson, L.P.; Pence, L.; Pinkas, G.; Song, H.; Telugu, B.P. Placental Hypoxia During Early Pregnancy Causes Maternal Hypertension and Placental Insufficiency in the Hypoxic Guinea Pig Model. Biol. Reprod. 2016, 95, 128.
- Gilbert, J.S.; Babcock, S.A.; Granger, J.P. Hypertension Produced by Reduced Uterine Perfusion in Fms-Like Tyrosine Kinase-1 Expression. Pregnancy Hypertens. 2007, 50, 1142–1147.
- Alexander, B.T.; Kassab, S.E.; Miller, M.T.; Abram, S.R.; Reckelhoff, J.F.; Bennett, W.A.; Granger, J.P. Reduced uterine perfusion pressure during pregnancy in the rat is associated with increases in arterial pressure and changes in renal nitric oxide. Hypertension 2001, 37, 1191–1195.
- Morton, J.S.; Levasseur, J.; Ganguly, E.; Quon, A.; Kirschenman, R.; Dyck, J.R.B. Characterisation of the Selective Reduced Uteroplacental Perfusion (sRUPP) Model of Preeclampsia. Sci. Rep. 2019, 9, 9565.
- Fushima, T.; Sekimoto, A.; Minato, T.; Ito, T.; Oe, Y.; Kisu, K.; Sato, E.; Funamoto, K.; Hayase, T.; Kimura, Y.; et al. Reduced uterine perfusion pressure (RUPP) model of preeclampsia in Mice. PLoS ONE 2016, 11, e0155426.
- Losonczy, G.; Brown, G.; Venuto, R.C. Reduced Uterine Perfusion Pressure Hypertension in Pregnant Rabbits. Am. J. Med. Sci. 1992, 303, 233–240.
- Maynard, S.E.; Min, J.Y.; Merchan, J.; Lim, K.H.; Li, J.; Mondal, S.; Libermann, T.A.; Morgan, J.P.; Sellke, F.W.; Stillman, I.E.; et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction hypertension, and proteinuria in preeclampsia. J. Clin. Investig. 2003, 111, 649–658.
- Venkatesha, S.; Toporsian, M.; Lam, C.; Hanai, J.; Mammoto, T.; Kim, Y.M.; Bdolah, Y.; Lim, K.; Yuan, H.; Libermann, T.A.; et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat. Med. 2006, 12, 642–650.
- Carlstrom, M.; Wentzel, P.; Skøtt, O.; Persson, A.E.G.; Carlstro, M.; Eriksson, U.J. Angiogenesis inhibition causes hypertension and placental dysfunction in a rat model of preeclampsia. J. Hypertens. 2009, 27, 829–837.
- Tal, R.; Shaish, A.; Barshack, I.; Polak-Charcon, S.; Afek, A.; Volkov, A.; Feldman, B.; Avivi, C.; Harats, D. Effects of hypoxia-inducible factor-1α overexpression in pregnant mice: Possible implications for preeclampsia and intrauterine growth restriction. Am. J. Pathol. 2010, 177, 2950–2962.
- Chang, X.; Yao, J.; He, Q.; Liu, M.; Duan, T.; Wang, K. Exosomes from Women with Preeclampsia Induced Vascular Dysfunction by Delivering sFlt (Soluble Fms-Like Tyrosine Kinase)-1 and sEng (Soluble Endoglin) to Endothelial Cells. Hypertension 2018, 72, 1381–1390.
- Bergmann, A.; Ahmad, S.; Cudmore, M.; Gruber, A.D.; Wittschen, P.; Gröne, H.; Ahmed, A.; Weich, H.A. Reduction of circulating soluble Flt-1 alleviates preeclampsia-like symptoms in a mouse model. J. Cell. Mol. Med. 2010, 14, 1857–1867.
- Lu, F.; Longo, M.; Tamayo, E.; Maner, W.; Al-hendy, A.; Anderson, G.D.; Hankins, G.D.V.; Saade, G.R. The effect of over-expression of sFlt-1 on blood pressure and the occurrence of other manifestations of preeclampsia in unrestrained conscious pregnant mice. Am. J. Obstet. Gynecol. 2007, 196, 396.e1–396.e7.
- Kumasawa, K.; Ikawa, M.; Kidoya, H.; Hasuwa, H.; Saito-Fujita, T.; Morioka, Y.; Takakura, N.; Kimura, T.; Okabe, M. Pravastatin induces placental growth factor (PGF) and ameliorates preeclampsia in a mouse model. Proc. Natl. Acad. Sci. USA 2011, 108, 1451–1455.
- Warrington, J.P.; Drummond, H.A.; Granger, J.P.; Ryan, M.J. Placental ischemia-induced increases in brain water content and cerebrovascular permeability: Role of TNF-α. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 309, R1425–R1431.
- George, E.M.; Stout, J.M.; Stec, D.E.; Granger, J.P. Heme oxygenase induction attenuates TNF-α-induced hypertension in pregnant rodents. Front. Pharmacol. 2015, 6, 165.
- LaMarca, B.; Speed, J.; Ray, L.F.; Cockrell, K.; Wallukat, G.; Dechend, R.; Granger, J. Hypertension in response to IL-6 during pregnancy: Role of AT1-receptor activation. Int. J. Interf. Cytokine Mediat. Res. 2011, 3, 65–70.
- Irani, R.A.; Zhang, Y.; Zhou, C.C.; Blackwell, S.C.; Hicks, M.J.; Ramin, S.M.; Kellems, R.E.; Xia, Y. Autoantibody-mediated angiotensin receptor activation contributes to preeclampsia through tumor necrosis factor-α signaling. Hypertension 2010, 55, 1246–1253.
- Bobek, G.; Surmon, L.; Mirabito, K.M.; Makris, A.; Hennessy, A. Placental Regulation of Inflammation and Hypoxia after TNF- a Infusion in Mice. Am. J. Reprod. Immunol. 2015, 74, 407–418.
- Chatterjee, P.; Chiasson, V.L.; Kopriva, S.E.; Young, K.J.; Chatterjee, V.; Jones, K.A.; Mitchell, B.M. Interleukin 10 deficiency exacerbates toll-like receptor 3-induced preeclampsia-like symptoms in mice. Hypertension 2011, 58, 489–496.
- Heyward, C.; Sones, J.L.; Lob, H.; Yuen, L.; Abbott, K.; Huang, W.; Begun, Z.; Butler, S.; August, A.; Davisson, R. The decidua of preeclamptic-like BPH/5 mi. J. Reprod. Immunol. 2017, 120, 27–33.
- Zuo, J.; Jiang, Z. Melatonin attenuates hypertension and oxidative stress in a rat model of L-NAME-induced gestational hypertension. Vasc. Med. 2020, 25, 295–301.
- Shu, W.E.N.; Li, H.; Gong, H.A.O.; Zhang, M.E.I.; Niu, X.; Ma, Y.; Zhang, X.I.N.; Cai, W.E.I.; Yang, G.; Wei, M.; et al. Evaluation of blood vessel injury, oxidative stress and circulating inflammatory factors in an L—NAME—Induced preeclampsia-like rat model. Exp. Ther. Med. 2018, 16, 585–594.
- Liu, F.; Wang, Y.; Wang, X.; Zheng, Y.; Jin, Z.; Zhi, J. Role of agonistic autoantibodies against type-1 angiotensin II receptor in the pathogenesis of retinopathy in preeclampsia. Sci. Rep. 2016, 6, 29036.
- Huai, J.; Yang, Z.; Yi, Y.H.; Wang, G.J. Different Effects of Pravastatin on Preeclampsia—Like Symptoms in Different Mouse Models. Chin. Med. J. 2018, 131, 461–470.
- Zhou, C.C.; Zhang, Y.; Irani, R.A.; Zhang, H.; Mi, T.; Popek, E.J.; Hicks, M.J.; Ramin, S.M.; Kellems, R.E.; Xia, Y. Angiotensin receptor agonistic autoantibodies induce pre-eclampsia in pregnant mice. Nat. Med. 2008, 14, 855–862.
- Kusinski, L.C.; Stanley, J.L.; Dilworth, M.R.; Hirt, C.J.; Andersson, I.J.; Renshall, L.J.; Baker, B.C.; Baker, P.N.; Sibley, C.P.; Wareing, M.; et al. eNOS knockout mouse as a model of fetal growth restriction with an impaired uterine artery function and placental transport phenotype. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 303, R86–R93.
- Pijnenborg, R.; Vercruysse, L. Animal models of deep trophoblast invasion. In Placental Bed Disorders: Basic Science and Its Translation to Obstetrics; Cambridge University Press: Cambridge, UK, 2010; pp. 127–139. ISBN 9780511750847.
- Xue, L.U.; Xie, K.; Wu, L.A.N.; Yu, X.; Long, W.E.I.; Li, C.; Jia, R.; Ding, H. A novel peptide relieves endothelial cell dysfunction in preeclampsia by regulating the PI3K/mTOR/HIF1 α pathway. Int. J. Mol. Med. 2021, 47, 276–288.
- Cotechini, T.; Komisarenko, M.; Sperou, A.; Macdonald-Goodfellow, S.; Adams, M.A.; Graham, C.H. Inflammation in rat pregnancy inhibits spiral artery remodeling leading to fetal growth restriction and features of preeclampsia. J. Exp. Med. 2014, 211, 165–179.
- LaMarca, B.D.; Ryan, M.J.; Granger, J.P. Pathophysiology of Hypertension Inflammatory Cytokines During Preeclampsia: Role of Inflammatory Cytokines. Curr. Hypertens. Rev. 2007, 3, 69–74.
- Boggess, K.A.; Berggren, E.K.; Koskenoja, V.; Urlaub, D.; Lorenz, C. Severe preeclampsia and maternal self-report of oral health, hygiene, and dental care. J. Periodontol. 2013, 84, 143–151.
- Varshney, S.; Gautam, A. Poor periodontal health as a risk factor for development of pre-eclampsia in pregnant women. J. Indian Soc. Periodontol. 2014, 18, 321–325.
- Zhuo, J.; Xiao, D.; Hu, Y.; Wang, Z.; Paradis, A.; MataGreenwood, E.; Zhang, L. Gestational hypoxia induces preeclampsia-like symptoms via heightened endothelin-1 signaling in pregnant rats. Hypertension 2013, 62, 599–607.
- Ren, Z.; Gao, Y.; Gao, Y.; Liang, G.; Chen, Q.; Jiang, S.; Yang, X.; Fan, C.; Wang, H.; Wang, J.; et al. Distinct placental molecular processes associated with early-onset and late-onset preeclampsia. Theranostics 2021, 11, 5028–5044.
- Phipps, E.; Prasanna, D.; Brima, W.; Jim, B. Preeclampsia: Updates in Pathogenesis, Definitions, and Guidelines. Clin. J. Am. Soc. Nephrol. 2016, 11, 1102–1113.
- Gathiram, P.; Moodley, J. Pre-eclampsia: Its pathogenesis and pathophysiolgy. Cardiovasc. J. Afr. 2016, 27, 71–78.
- Marshall, S.A.; Hannan, N.J.; Jelinic, M.; Nguyen, T.P.H.; Girling, J.E.; Parry, L.J. Animal models of preeclampsia: Translational failings and why. Am. J. Physiol. Integr. Comp. Physiol. 2018, 314, R499–R508.
- Yart, L.; Roset Bahmanyar, E.; Cohen, M.; Martinez de Tejada, B. Role of the Uteroplacental Renin-Angiotensin System in Placental Development and Function, and Its Implication in the Preeclampsia Pathogenesis. Biomedicines 2021, 9, 1332.
- Rodriguez, M.; Moreno, J.; Hasbun, J. RAS in Pregnancy and Preeclampsia and Eclampsia. Int. J. Hypertens. 2012, 2012, 739274.
- Procopciuc, L.M.; Nemeti, G.; Buzdugan, E.; Iancu, M.; Stamatian, F.; Caracostea, G. Renin-angiotensin system gene variants and risk of early- and late-onset preeclampsia: A single center case-control study. Pregnancy Hypertens. 2019, 18, 1–8.
- Li, J.; LaMarca, B.; Reckelhoff, J.F. A model of preeclampsia in rats: The reduced uterine perfusion pressure (RUPP) model. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H1–H8.
- Saif, J.; Ahmad, S.; Rezai, H.; Litvinova, K.; Sparatore, A.; Alzahrani, F.A.; Wang, K.; Ahmed, A. Redox Biology Hydrogen sulfide releasing molecule MZe786 inhibits soluble Flt-1 and prevents preeclampsia in a refined RUPP mouse model. Redox Biol. 2021, 38, 101814.
- Abitbol, M.M. Simplified technique to produce toxemia in the rat: Consideration on cause of toxemia. Clin. Exp. Hypertens. Hypertens. Pregnancy 1982, 1, 93–103.
- Podjarny, E.; Losonczy, G.; Baylis, C. Animal Models of Preeclampsia. Semin. Nephrol. 2004, 24, 596–606.
- Zhu, M.; Ren, Z.; Possomato-vieira, J.S.; Khalil, R.A. Restoring placental growth factor-soluble fms-like tyrosine kinase-1 balance reverses vascular hyper-reactivity and hypertension in pregnancy. Am. J. Physiol. Integr. Comp. Physiol. 2016, 311, R505–R521.
- Gutkowska, J.; Granger, J.P.; LaMarca, B.B.; Danalache, B.A.; Wang, D.; Jankowski, M. Changes in cardiac structure in hypertension produced by placental ischemia in pregnant rats: Effect of tumor necrosis factor blockade. J. Hypertens. 2011, 29, 1203–1212.
- Gadonski, G.; LaMarca, B.B.D.; Sullivan, E.; Bennett, W.; Chandler, D.; Granger, J.P. Pregnancy and IL-6 Hypertension Produced by Reductions in Uterine Perfusion in the Pregnant Rat Role of Interleukin 6. Hypertension 2006, 48, 711–716.
- Waker, C.A.; Kaufman, M.R.; Brown, T.L. Current State of Preeclampsia Mouse Models: Approaches, Relevance, and Standardization. Front. Physiol. 2021, 12, 681632.
- LaMarca, B.; Speed, J.; Fournier, L.; Babcock, S.A.; Berry, H.; Granger, J.P. Hypertension in Response to Chronic Reductions in Uterine Perfusion in Pregnant Rats: Effect of Tumor Necrosis Factor-α Blockade. Hypertension 2008, 52, 1161–1167.
- Opichka, M.A.; Rappelt, M.W.; Gutterman, D.D.; Grobe, J.L.; McIntosh, J.J. Review vascular dysfunction in preeclampsia. Cells 2021, 10, 3055.
- Bridges, J.P.; Gilbert, J.S.; Colson, D.; Gilbert, S.A.; Dukes, M.P.; Ryan, M.J.; Granger, J.P. Oxidative stress contributes to soluble fms-like tyrosine kinase-1 induced vascular dysfunction in pregnant rats. Am. J. Hypertens. 2009, 22, 564–568.
- Szalai, G.; Romero, R.; Chaiworapongsa, T.; Xu, Y.; Wang, B.; Ahn, H.; Xu, Z.; Chiang, P.J.; Sundell, B.; Wang, R.; et al. Full-Length Human Placental sFlt-1-e15a Isoform Induces Distinct Maternal Phenotypes of Preeclampsia in Mice. PLoS ONE 2015, 10, e0119547.
- Murphy, S.R.; LaMarca, B.; Cockrell, K.; Arany, M.; Granger, J.P. L-Arginine supplementation abolishes the blood pressure and endothelin response to chronic increases in plasma sFlt-1 in pregnant rats. Am. J. Physiol. Integr. Comp. Physiol. 2012, 302, 259–263.
- Turanov, A.A.; Lo, A.; Hassler, M.R.; Makris, A.; Ashar-patel, A.; Alterman, J.F.; Coles, A.H.; Haraszti, R.A.; Roux, L.; Godinho, B.M.D.C.; et al. Articles RNAi modulation of placental sFLT1 for the treatment of preeclampsia. Nat. Biotechnol. 2018, 36, 1164–1175.
- Li, Z.; Zhang, Y.; Ma, J.Y.; Kapoun, A.M.; Shao, Q.; Kerr, I.; Lam, A.; Young, G.O.; Sannajust, F.; Stathis, P.; et al. Recombinant Vascular Endothelial Growth Factor 121 Attenuates Hypertension and Improves Kidney Damage in a Rat Model of Preeclampsia. Hypertension 2007, 50, 686–692.
- Eddy, A.C.; Iii, G.L.B.; George, E.M. Pro-angiogenic therapeutics for preeclampsia. Biol. Sex Differ. 2018, 9, 36.
- Matsubara, K.; Matsubara, Y.; Uchikura, Y.; Sugiyama, T. Pathophysiology of Preeclampsia: The Role of Exosomes. Int. J. Mol. Sci. 2021, 22, 2572.
- Solomon, C.G.; Seely, E.W. Preeclampsia—Searching for the Cause. N. Engl. J. Med. 2004, 350, 641–642.
- Murphy, S.R.; LaMarca, B.B.D.; Parrish, M.; Cockrell, K.; Granger, J.P. Control of soluble fms-like tyrosine-1 (sFlt-1) production response to placental ischemia/hypoxia: Role of tumor necrosis factor-α. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 304, R130–R135.
- Philogene, M.C.; Han, D.; Alvarado, F.; Fedarko, N.S.; Zonderman, A.B.; Evans, M.K.; Crews, D.C. Prevalence of Angiotensin II Type 1 Receptor Antibodies in Persons with Hypertension and Relation to Blood Pressure and Medication. Am. J. Hypertens. 2020, 33, 734–740.
- Campbell, N.; LaMarca, B.; Cunningham, M.W.J. The role of Agonistic Autoantibodies to the Angiotensin II Type 1 Receptor (AT1-AA) in Pathophysiology of Preeclampsia. Curr. Pharm. Biotechnol. 2018, 19, 781–785.
- LaMarca, B.D.; Cockrell, K.; Sullivan, E.; Bennett, W.; Granger, J.P. Role of Endothelin in Mediating Tumor Necrosis Factor-Induced Hypertension in Pregnant Rats. Hypertension 2005, 46, 82–87.
- Giardina, J.B.; Green, G.M.; Cockrell, K.L.; Granger, J.P.; Khalil, R.A.; Jena, B.; Green, G.M.; Kathy, L.; Granger, J.P.; Tnf-, R.A.K. TNF-a enhances contraction and inhibits endothelial NO-cGMP relaxation in systemic vessels of pregnant rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 283, R130–R143.
- Shesely, E.G.; Gilbert, C.; Granderson, G.; Carretero, C.D.; Carretero, O.A.; Beierwaltes, W.H. Nitric oxide synthase gene knockout mice do not become hypertensive during pregnancy. Am. J. Obstet. Gynecol. 2001, 185, 1198–1203.
- Kulandavelu, S.; Whiteley, K.J.; Qu, D.; Mu, J.; Bainbridge, S.A.; Adamson, S.L. Endothelial nitric oxide synthase deficiency reduces uterine blood flow, spiral artery elongation, and placental oxygenation in pregnant mice. Hypertension 2012, 60, 231–238.
- Nathan, L.; Cuevas, J.; Chaudhuri, G. The role of nitric oxide in the altered vascular reactivity of pregnancy in the rat. Br. J. Pharmacol. 1995, 114, 955–960.
- Ma, X.P.; Liu, C.D.; Cao, G.M.; Zhang, Z.Y. Transthyretin increases migration and invasion of rat placental trophoblast cells. FEBS Openbio 2020, 10, 1568–1576.
- Zhu, H.; Zhu, W.; Hu, R.; Wang, H.; Ma, D.; Li, X. The effect of pre-eclampsia-like syndrome induced by L-NAME on learning and memory and hippocampal glucocorticoid receptor expression: A rat model. Hypertens. Pregnancy 2017, 36, 36–43.
- De Alwis, N.; Binder, N.K.; Beard, S.; Mangwiro, Y.T.M.; Kadife, E.; Cuffe, J.S.M.; Keenan, E.; Fato, B.R.; Kaitu, T.J.; Brownfoot, F.C.; et al. The L-NAME mouse model of preeclampsia and impact to long-term maternal cardiovascular health. Life Sci. Alliance 2022, 5, e202201517.
- Losonczy, G.; Mucha, I.; Muller, V.; Kriston, T.; Ungvari, Z.; Tornoci, L.; Rosivall, L.; Venuto, R. The vasoconstrictor effects of L-NAME, a nitric oxide synthase inhibitor, in pregnant rabbits. Br. J. Pharmacol. 1996, 118, 1012–1018.
- Yoshikawa, K.; Umekawa, T.; Maki, S.; Kubo, M.; Nii, M.; Tanaka, K.; Tanaka, H.; Osato, K.; Kamimoto, Y.; Kondo, E.; et al. Tadalafil Improves L-NG-Nitroarginine Methyl Ester-Induced Preeclampsia with Fetal Growth Restriction-Like Symptoms in Pregnant Mice. Am. J. Hypertens. 2018, 31, 89–96.
- Selivanova, E.K.; Shvetsova, A.A.; Borzykh, A.A.; Gaynullina, D.K.; Kiryukhina, O.O.; Lukoshkova, E.V.; Potekhina, V.M.; Kuzmin, V.S.; Tarasova, O.S. Intrauterine L-NAME Exposure Weakens the Development of Sympathetic Innervation and Induces the Remodeling of Arterial Vessels in Two-Week-Old Rats. Int. J. Mol. Med. 2021, 22, 12327.
- Teng, R.; Wu, T.; Sharma, R.; Garrison, R.D.; Hudak, M.L. Early neonatal hypotension in premature infants born to preeclamptic mothers. J. Perinatol. 2006, 26, 471–475.
- Danielson, L.A.; Conrad, K.P. Acute Blockade of Nitric Oxide Synthase Inhibits Renal Vasodilation and Hyperfiltration During Pregnancy in Chronically Instrumented Conscious Rats. J. Clin. Investig. 1995, 96, 482–490.
- Seligman, S.P.; Buyon, J.P.; Clancy, R.M.; Young, B.K.; Abramson, S.B. The role of nitric oxide in the pathogenesis of preeclampsia. Am. J. Obstet. Gynecol. 1994, 171, 944–948.
- Cushen, S.C.; Goulopoulou, S. New Models of Pregnancy-Associated Hypertension. Am. J. Hypertens. 2017, 30, 1053–1062.
More
Information
Subjects:
Obstetrics & Gynaecology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.0K
Revisions:
2 times
(View History)
Update Date:
27 Dec 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No