Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sonja Vermeren | -- | 2153 | 2022-12-22 03:37:08 | | | |
| 2 | Conner Chen | + 3 word(s) | 2156 | 2022-12-22 05:23:20 | | | | |
| 3 | Conner Chen | + 24 word(s) | 2180 | 2022-12-30 03:34:36 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Loh, W.; Vermeren, S. Neutrophil Apoptosis as A Powerful Anti-Inflammatory Signal. Encyclopedia. Available online: https://encyclopedia.pub/entry/39063 (accessed on 04 August 2026).
Loh W, Vermeren S. Neutrophil Apoptosis as A Powerful Anti-Inflammatory Signal. Encyclopedia. Available at: https://encyclopedia.pub/entry/39063. Accessed August 04, 2026.
Loh, Waywen, Sonja Vermeren. "Neutrophil Apoptosis as A Powerful Anti-Inflammatory Signal" Encyclopedia, https://encyclopedia.pub/entry/39063 (accessed August 04, 2026).
Loh, W., & Vermeren, S. (2022, December 22). Neutrophil Apoptosis as A Powerful Anti-Inflammatory Signal. In Encyclopedia. https://encyclopedia.pub/entry/39063
Loh, Waywen and Sonja Vermeren. "Neutrophil Apoptosis as A Powerful Anti-Inflammatory Signal." Encyclopedia. Web. 22 December, 2022.
Copy Citation
Neutrophils are highly abundant circulating leukocytes that are amongst the first cells to be recruited to sites of infection or sterile injury. Their ability to generate and release powerful cytotoxic products ties with their role in host defence from bacterial and fungal infections. Neutrophilic inflammation is tightly regulated to limit the amount of ‘bystander injury’ caused. Neutrophils were in the past regarded as short-lived, indiscriminate killers of invading microorganisms. Neutrophils are recognised to also have important anti-inflammatory functions that are critical for the resolution of inflammation and return to homeostasis.
neutrophil
macrophage
inflammation
apoptosis
neutrophil extracellular trap
1. Neutrophil Apoptosis—A Powerful Anti-Inflammatory Signal
Neutrophil apoptosis is triggered in aged neutrophils as part of homeostasis, but it may also be induced in specific situations. For example, phagocytosis and intracellular killing of serum or antibody-opsonised pathogens induces a specialised cellular differentiation programme that culminates in phagocytosis-induced cell death (PICD) ([1][2][3][4] and see reactive oxygen species (ROS) below). Given the anti-inflammatory nature of neutrophil apoptosis, this mechanism is perfectly suited for promoting tissue repair once the infection has been cleared.
In the absence of pathogens, insoluble antibody complexes, powerful pro-inflammatory stimuli that are abundant in biological fluids in autoimmune disease (e.g., the synovial fluid in rheumatoid arthritis), are internalised by neutrophils by receptor-dependent macropinocytosis [5]. Using a separate pathway, the immune complexes also promote ROS-dependent neutrophil apoptosis. Immune complex-induced neutrophil apoptosis is mechanistically distinct from PICD [5][6][7]. This anti-inflammatory mechanism is likely relevant for immune complex-driven autoimmune diseases, such as rheumatoid arthritis and lupus [8]. In addition, it may be involved in clearing excess antibody and promoting repair processes after infectious diseases, when rheumatoid factor, an Fc region binding IgM, is temporarily expressed, generating large circulating immune complexes [9][10].
2. Apoptotic Neutrophils Generate ‘Find-Me’ and ‘Eat-Me’ Signals to Promote Efferocytosis and Macrophage Polarisation
Irrespective of its mode of induction, by maintaining the integrity of its plasma membrane, neutrophil apoptosis offers a window of opportunity for clearance of the dead cells without eliciting inflammation. To this end, apoptotic neutrophils promote their own clearance by macrophages via a specialised phagocytosis mechanism known as efferocytosis (Figure 1; reviewed in [11][12]). Efferocytosis not only prevents secondary necrosis of the apoptotic neutrophils; it is also anti-inflammatory, promotes pro-resolving pathways, and initiates repair.
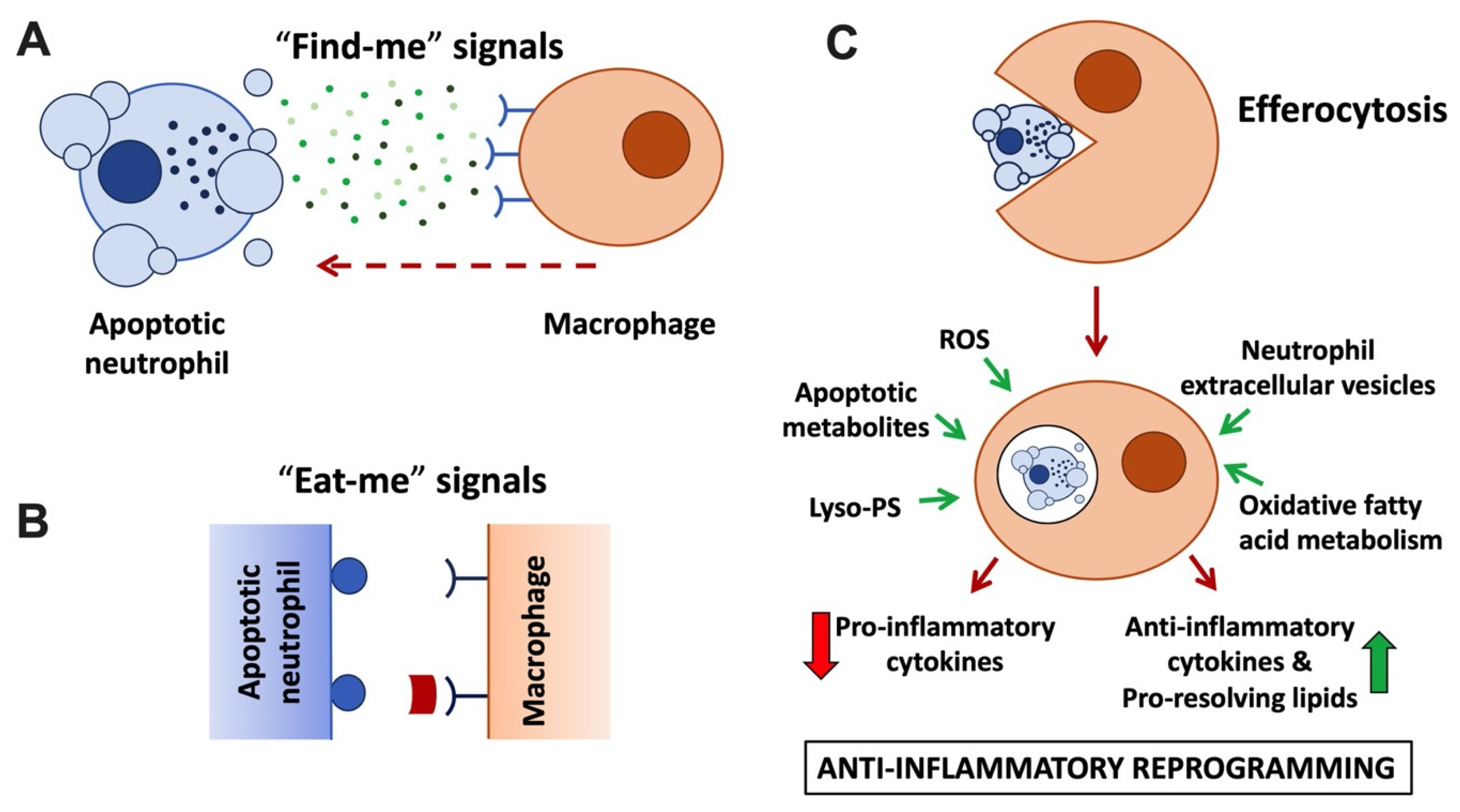
Figure 1. Anti-inflammatory neutrophil functions linked to apoptosis and efferocytosis. (A) Apoptotic neutrophils and their extracellular vesicles release ‘find-me’ signals, including nucleotides (ATP, UTP), lysophosphatidylcholine, and sphingosine-1-phosphate to attract macrophages. ‘Find-me’ signals bind to cognate receptors on macrophages. (B) Apoptotic neutrophils and blebs display ‘eat-me’ signals on their plasma membrane. These include phosphatidylserine (PS) and lyso-PS, which are recognised by macrophage receptors (e.g., BAI1 and TIM family) or that are recognised via bridging molecules, such as protein S and Gas6 that bind to TAM family macrophage receptors. (C) Neutrophil efferocytosis. Macrophages engulf apoptotic neutrophils and digest them in an immunologically silent fashion in an efferosome or LAPosome. Released apoptotic cell and fatty acid metabolites ligate metabolite sensing receptors (LXR, PPAR), upregulating transcription of anti-inflammatory cytokines and potentiating efferocytosis. Other signals promoting macrophage reprogramming to the anti-inflammatory phenotype (also known as M2) include ROS, lyso-PS, and neutrophil extracellular vesicles.
3. ‘Find-Me’ Signals
Recognition of apoptotic cells by macrophages is promoted via soluble mediators, or ‘find-me’ signals, that are actively released by apoptotic cells and/or extracellular vesicles (Figure 1A; reviewed by [13]). Low concentrations of nucleotides ATP and UTP released by apoptotic neutrophils act as ‘find-me’ signals that bind to the macrophage P2Y2 purinergic receptor [14], while the lipid mediators lysophosphatidylcholine (LPC) and sphingosine 1-phosphate bind to macrophage G2A and S1P1-5 receptors, respectively [15][16]. Besides acting as chemoattractants, ‘find-me’ signals were also reported to have immunomodulatory functions, enhancing the phagocytic activity of macrophages, and mediating anti-inflammatory effects by modulating cytokine production [17][18][19], although some of the underlying mechanisms remain to be fully explored. A recent metabolomics analysis performed with a range of apoptotic immune cells suggested that shared core metabolites act in a cooperative fashion as ‘find-me’ signals [20].
4. ‘Eat-Me’ Signals
Apoptotic cells also display prominent ‘eat-me’ signals on their surface. These ‘eat-me’ signals are recognised by specialised phagocytic receptors (Figure 1B) and trigger internalisation of the apoptotic corpse. The most prominent ‘eat-me’ signal is the display of the membrane lipid phosphatidylserine (PS) on the outside of the cell. Viable cells are characterised by phospholipid asymmetry. This is due to flippase transporters, which actively limit PS to the inner leaflet of the plasma membrane. In apoptotic cells, flippases are inactivated in a caspase-dependent fashion; simultaneously, scramblases are activated, further disturbing PS asymmetry [21][22][23]. Recognition of PS can be direct via PS receptors, including BAI1 or TIM (T cell/transmembrane, immunoglobulin, and mucin) family receptors. In addition, Tyro/Axl/Mer (TAM) family receptors recognise PS indirectly via soluble bridging molecules, such as protein S and Gas6 (reviewed by [24]). Interestingly, in addition to enabling efferocytosis, TAM receptor activation was reported to elicit additional anti-inflammatory functions, suppressing inflammatory cytokine production and promoting macrophage polarisation toward an alternative or M2-like phenotype [25][26]. A number of ‘eat-me’ signals other than PS exist, e.g., calreticulin and pentraxin 3; these may be expressed in a cell type-specific fashion and act to further promote efferocytosis alongside PS.
In contrast with apoptotic neutrophils, viable cells display ‘do not eat-me signals’ on their surface which interfere with their uptake by macrophages by efferocytosis. ‘Do not eat-me signals’ even counteract the internalisation of PS-displaying cells [27]. For example, the ‘do not eat-me’ signal CD47 binds to its macrophage receptor SIRPα, interfering with the internalisation of bound cells by employing the phosphatase SHP1/2 (for details see review [28]).
5. Efferocytosis
Following uptake of the apoptotic neutrophil by the macrophage by efferocytosis, degradation of the apoptotic cell ensues (Figure 1C). Degradation was reported to occur by a Rab5, Rab7, and Rab17-dependent variant of the traditional endocytic pathway in an immunologically silent fashion [29][30]. Separately, efferocytosis was also reported to occur via an LC3-associated phagocytosis pathway, short LAP, a non-canonical form of autophagy [31]. LAP stands in sharp contrast to the pro-inflammatory degradative pathway by which degradation of microbes occurs following phagocytosis. LAP employs the phagocyte’s autophagic machinery to degrade the internalised apoptotic material in an anti-inflammatory fashion. Instead of generating an autophagosome, in LAP, LC3 is conjugated to the phagosome or ‘LAPosome’ [31][32]. LAP-deficient animals accumulate apoptotic bodies in their tissues that are reminiscent of the dead cells found in the circulation of systemic lupus erythematosus (SLE) patients, and genetic links between autoimmunity and defective LAP have been identified [33][34]. As with classical autophagy, LAP ultimately results in the release of apoptotic metabolites into the macrophage; it upregulates fatty acid oxidation, which has been proposed to be critical for the anti-inflammatory nature of efferocytosis.
6. Neutrophil-Dependent Macrophage Reprogramming
Efferocytosis has long been regarded as non-inflammatory. This is (i) because it avoids the release of neutrophil granule contents, limiting tissue injury [35], and (ii) because uptake of apoptotic neutrophils by macrophages was observed to induce their production of anti-inflammatory cytokines (e.g., TGF-β and IL-10) and pro-resolving lipid species (resolvins D1, D2, E4, lipoxin A4 and maresin) at the expense of pro-inflammatory cytokines (e.g., TNF, IL-1β and IL-12) (Figure 1C; [36][37][38]). This stands in sharp contrast to the pro-inflammatory cytokine profile observed after phagocytosis of microbes and nurtured the notion that efferocytosis promotes macrophage polarisation towards a pro-resolution phenotype (also referred to as M2). While the mechanism underpinning the switch from pro- to anti-inflammatory cytokines has not yet been elucidated in its entirety, recent studies have suggested an involvement of metabolic modulation of macrophages post-efferocytosis in conjunction with metabolite sensing nuclear receptors to fine-tune anti-inflammatory processes and regulate the transcription of pro- and anti-inflammatory cytokines. Apoptotic cell-derived metabolites were shown to drive enhanced transcription of engulfment-related genes, including PS receptors and bridging molecules, further increasing macrophage efferocytosis capacity. In addition, genetic experiments identified roles of nuclear fatty acid liver X receptors (LXR) and peroxisome-proliferator-activated receptors (PPAR) for upregulation of TGF-β and IL-10, and the suppression of TNF, IL-1β, and IL-12 [39][40][41]. Recent elegant studies identified that oxidative fatty acid metabolism specifically promotes IL-10 production by macrophages [42]. Additionally, amino acids obtained from apoptotic cells, such as arginine, are metabolised promoting the long-term potentiation of efferocytosis and contributing to the resolution of inflammation [43].
7. Neutrophil Extracellular Vesicles
In recent years, extracellular vesicles (EVs) have emerged as a means by which cells communicate with one another in a range of situations. Neutrophilic EVs (sometimes also referred to as microvesicles or ectosomes) are produced by membrane blebbing (Figure 1) in response to a range of stimuli, including chemokines, cytokines, bacteria, and bacterial products at sites of inflammation. They contain membrane receptors, surface proteins, including the anti-inflammatory annexin-1 and PS, as well as active neutrophilic enzymes [44][45][46]. Although neutrophilic inflammatory EVs were reported to be generated under certain conditions [47][48], in many other studies, EVs generated by living or apoptotic neutrophils were shown to limit excessive inflammation in their surroundings. EVs are reported to orchestrate anti-inflammatory reprogramming of macrophages, reducing production of pro-inflammatory cytokines and increasing production of pro-resolving cytokines and lipid mediators, as well as their ability to efferocytose [45][49][50]. Indeed, neutrophilic EVs were shown to dampen inflammation, even in chronic inflammation, such as the rheumatic joint, acting on a range of cell types to protect cartilage [51][52]. Mechanistically, the ability of neutrophil EV to reprogramme macrophages relies on their exposure of PS and annexin-1. Annexin-1 binding to the macrophage FRP2 receptor was shown to be required for TGF-β expression. In contrast, PS exposure by EVs modulated macrophage expression of IL-1β, IL10, and IL-12. PS-binding to macrophage MerTK was moreover required to enhance the efferocytic activity of macrophages [52]. One recent study reported macrophage reprogramming by living or apoptotic neutrophils, even in the absence of efferocytosis, and identified that this involved transcriptional reprogramming of the macrophages and suppression of NF-κB activation [53]. However, it remains to be formally established whether this observation was indeed due to neutrophil EVs.
8. Reactive Oxygen Species (ROS)
Due to their essential function in microbial killing by neutrophils, ROS are vital for host immunity, but excessive ROS generation can drive oxidative damage to the host; for this reason, the generation of ROS is tightly controlled. Key to the generation of ROS is the activation of the NADPH oxidase, which occurs in a strictly regulated fashion that involves phosphoinositide-driven translocation events, as well as protein phosphorylation. The NADPH oxidase is assembled at a membrane (typically either phagosomal or plasma membrane); it is made up of membrane associated (gp91phox and p22phox) and cytosolic components (p67phox, p47phox, p40phox) in the presence of activated (GTP-loaded) Rac which binds p67phox (reviewed in [54]). Once assembled, the NADPH oxidase transfers NADPH-derived electrons to molecular oxygen, generating superoxide on the outside of the cell (which may be the inside of the phagosome). Further enzymatic reactions that are less tightly controlled generate increasingly more cytotoxic species, culminating in the generation of hypochlorous acid from hydrogen peroxide by myeloperoxidase, which is delivered from primary/azurophilic granules [54]. Missense mutations in any one of the subunits of the NADPH oxidase affect its function in human and mouse, causing chronic granulomatous disease (CGD) in humans [55]. Fascinatingly, ROS also have anti-inflammatory functions, at least some of which are due to their key role in apoptosis. Phagocytosis of microorganisms triggers activation of the NADPH oxidase, which in turn promotes PICD ([1][56] and Figure 1B). The same observation applies to immune complex-induced neutrophil apoptosis [5]. Experimental evidence suggests that the NADPH oxidase promotes efferocytosis of apoptotic PS-exposing neutrophils for the optimal resolution of inflammation. NADPH oxidase-dependent oxidation of the fatty acyl groups of PS culminated in the generation of lyso-PS, generating improved ligands for PS receptors, including CD36 and G2A [57][58]. Interestingly, ROS-dependent lyso-PS generated by viable neutrophils even promoted efferocytosis of these non-apoptotic cells, suggesting a potential role of lyso-PS in orchestrating the removal of excess neutrophils following their recruitment to inflammatory sites [59].
In keeping with these observations, mice lacking NADPH oxidase components were found to be prone to acute hyperinflammatory reactions in sterile inflammation models, exhibiting excessive inflammatory cell infiltrates and pro-inflammatory cytokine signatures (e.g., [60][61]). Failure of prompt efferocytosis of apoptotic cells promoted not only acute inflammation, but, in the long run, development of autoimmune disease, including SLE [62]. Genome-wide association studies identified patients carrying NADPH oxidase subunit mutations that reduced its activity in SLE cohorts (e.g., [33][63]). These observations also held true in mouse models [64][65][66]. Efferocytosis of apoptotic neutrophils by CGD macrophages, or those from CGD mouse models, was also reported to be affected: CGD macrophages were found to suffer from poor efferosome acidification and delayed clearance of ingested apoptotic cells [67]. Impaired efferocytosis resulted in poor macrophage reprogramming from pro-inflammatory (M1) to pro-resolving (M2), and reduced generation of anti-inflammatory mediators, including TGF-β and prostaglandin D2 (PGD2) [68][69][70]. In this regard, it is also interesting that, in the context of liver repair after acute acetaminophen (paracetamol) injury in the mouse, H2O2 released by neutrophils was sufficient to promote inflammatory monocyte differentiation to pro-repair macrophages by activating AMPK [71], even in the absence of neutrophil apoptosis and efferocytosis.
Taken together, these observations suggest important anti-inflammatory roles of NADPH oxidase-mediated generation of ROS, that counterintuitively suppress excessive bystander host injury from being generated due to neutrophilic inflammation.
References
- Watson, R.W.; Redmond, H.P.; Wang, J.H.; Condron, C.; Bouchier-Hayes, D. Neutrophils undergo apoptosis following ingestion of Escherichia coli. J. Immunol. 1996, 156, 3986–3992.
- Perskvist, N.; Long, M.; Stendahl, O.; Zheng, L. Mycobacterium tuberculosis promotes apoptosis in human neutrophils by activating caspase-3 and altering expression of Bax/Bcl-xL via an oxygen-dependent pathway. J. Immunol. 2002, 168, 6358–6365.
- Zhang, B.; Hirahashi, J.; Cullere, X.; Mayadas, T.N. Elucidation of molecular events leading to neutrophil apoptosis following phagocytosis: Cross-talk between caspase 8, reactive oxygen species, and MAPK/ERK activation. J. Biol. Chem. 2003, 278, 28443–28454.
- DeLeo, F.R. Modulation of phagocyte apoptosis by bacterial pathogens. Apoptosis 2004, 9, 399–413.
- Karmakar, U.; Chu, J.Y.; Sundaram, K.; Astier, A.L.; Garside, H.; Hansen, C.G.; Dransfield, I.; Vermeren, S. Immune complex-induced apoptosis and concurrent immune complex clearance are anti-inflammatory neutrophil functions. Cell Death Dis. 2021, 12, 296.
- Gamberale, R.; Giordano, M.; Trevani, A.S.; Andonegui, G.; Geffner, J.R. Modulation of human neutrophil apoptosis by immune complexes. J. Immunol. 1998, 161, 3666–3674.
- Chu, J.Y.; Dransfield, I.; Rossi, A.G.; Vermeren, S. Non-canonical PI3K-Cdc42-Pak-Mek-Erk Signaling Promotes Immune-Complex-Induced Apoptosis in Human Neutrophils. Cell Rep. 2016, 17, 374–386.
- Karmakar, U.; Vermeren, S. Crosstalk between B cells and neutrophils in rheumatoid arthritis. Immunology 2021, 164, 689–700.
- Hogben, D.N.; Devey, M.E. Studies on rheumatoid factor: I. The effect of rheumatoid factor on the clearance of preformed immune complexes in mice. Clin. Exp. Immunol. 1986, 66, 648–653.
- Newkirk, M.M. Rheumatoid factors: Host resistance or autoimmunity? Clin. Immunol. 2002, 104, 1–13.
- Kourtzelis, I.; Hajishengallis, G.; Chavakis, T. Phagocytosis of Apoptotic Cells in Resolution of Inflammation. Front. Immunol. 2020, 11, 553.
- Doran, A.C.; Yurdagul, A., Jr.; Tabas, I. Efferocytosis in health and disease. Nat. Rev. Immunol. 2020, 20, 254–267.
- Medina, C.B.; Ravichandran, K.S. Do not let death do us part: ‘find-me’ signals in communication between dying cells and the phagocytes. Cell Death Differ. 2016, 23, 979–989.
- Elliott, M.R.; Chekeni, F.B.; Trampont, P.C.; Lazarowski, E.R.; Kadl, A.; Walk, S.F.; Park, D.; Woodson, R.I.; Ostankovich, M.; Sharma, P.; et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 2009, 461, 282–286.
- Lauber, K.; Bohn, E.; Krober, S.M.; Xiao, Y.J.; Blumenthal, S.G.; Lindemann, R.K.; Marini, P.; Wiedig, C.; Zobywalski, A.; Baksh, S.; et al. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell 2003, 113, 717–730.
- Gude, D.R.; Alvarez, S.E.; Paugh, S.W.; Mitra, P.; Yu, J.; Griffiths, R.; Barbour, S.E.; Milstien, S.; Spiegel, S. Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a “come-and-get-me” signal. FASEB J. 2008, 22, 2629–2638.
- Koizumi, S.; Shigemoto-Mogami, Y.; Nasu-Tada, K.; Shinozaki, Y.; Ohsawa, K.; Tsuda, M.; Joshi, B.V.; Jacobson, K.A.; Kohsaka, S.; Inoue, K. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature 2007, 446, 1091–1095.
- Marques-da-Silva, C.; Burnstock, G.; Ojcius, D.M.; Coutinho-Silva, R. Purinergic receptor agonists modulate phagocytosis and clearance of apoptotic cells in macrophages. Immunobiology 2011, 216, 1–11.
- Yamaguchi, H.; Maruyama, T.; Urade, Y.; Nagata, S. Immunosuppression via adenosine receptor activation by adenosine monophosphate released from apoptotic cells. Elife 2014, 3, e02172.
- Medina, C.B.; Mehrotra, P.; Arandjelovic, S.; Perry, J.S.A.; Guo, Y.; Morioka, S.; Barron, B.; Walk, S.F.; Ghesquiere, B.; Krupnick, A.S.; et al. Metabolites released from apoptotic cells act as tissue messengers. Nature 2020, 580, 130–135.
- Suzuki, J.; Denning, D.P.; Imanishi, E.; Horvitz, H.R.; Nagata, S. Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science 2013, 341, 403–406.
- Segawa, K.; Nagata, S. An Apoptotic ‘Eat Me’ Signal: Phosphatidylserine Exposure. Trends Cell Biol. 2015, 25, 639–650.
- Fujii, T.; Sakata, A.; Nishimura, S.; Eto, K.; Nagata, S. TMEM16F is required for phosphatidylserine exposure and microparticle release in activated mouse platelets. Proc. Natl. Acad. Sci. USA 2015, 112, 12800–12805.
- Barth, N.D.; Marwick, J.A.; Vendrell, M.; Rossi, A.G.; Dransfield, I. The “Phagocytic Synapse” and Clearance of Apoptotic Cells. Front. Immunol. 2017, 8, 1708.
- Filardy, A.A.; Pires, D.R.; Nunes, M.P.; Takiya, C.M.; Freire-de-Lima, C.G.; Ribeiro-Gomes, F.L.; DosReis, G.A. Proinflammatory clearance of apoptotic neutrophils induces an IL-12(low)IL-10(high) regulatory phenotype in macrophages. J. Immunol. 2010, 185, 2044–2050.
- Zizzo, G.; Hilliard, B.A.; Monestier, M.; Cohen, P.L. Efficient clearance of early apoptotic cells by human macrophages requires M2c polarization and MerTK induction. J. Immunol. 2012, 189, 3508–3520.
- Segawa, K.; Suzuki, J.; Nagata, S. Constitutive exposure of phosphatidylserine on viable cells. Proc. Natl. Acad. Sci. USA 2011, 108, 19246–19251.
- Kelley, S.M.; Ravichandran, K.S. Putting the brakes on phagocytosis: “don’t-eat-me” signaling in physiology and disease. EMBO Rep. 2021, 22, e52564.
- Kinchen, J.M.; Doukoumetzidis, K.; Almendinger, J.; Stergiou, L.; Tosello-Trampont, A.; Sifri, C.D.; Hengartner, M.O.; Ravichandran, K.S. A pathway for phagosome maturation during engulfment of apoptotic cells. Nat. Cell Biol. 2008, 10, 556–566.
- Yin, C.; Kim, Y.; Argintaru, D.; Heit, B. Rab17 mediates differential antigen sorting following efferocytosis and phagocytosis. Cell Death Dis. 2016, 7, e2529.
- Martinez, J.; Almendinger, J.; Oberst, A.; Ness, R.; Dillon, C.P.; Fitzgerald, P.; Hengartner, M.O.; Green, D.R. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc. Natl. Acad. Sci. USA 2011, 108, 17396–17401.
- Sanjuan, M.A.; Dillon, C.P.; Tait, S.W.; Moshiach, S.; Dorsey, F.; Connell, S.; Komatsu, M.; Tanaka, K.; Cleveland, J.L.; Withoff, S.; et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 2007, 450, 1253–1257.
- The International Consortium for Systemic Lupus Erythematosus Genetics (SLEGEN); Harley, J.B.; Alarcon-Riquelme, M.E.; Criswell, L.A.; Jacob, C.O.; Kimberly, R.P.; Moser, K.L.; Tsao, B.P.; Vyse, T.J.; Langefeld, C.D. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat. Genet. 2008, 40, 204–210.
- Gateva, V.; Sandling, J.K.; Hom, G.; Taylor, K.E.; Chung, S.A.; Sun, X.; Ortmann, W.; Kosoy, R.; Ferreira, R.C.; Nordmark, G.; et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat. Genet. 2009, 41, 1228–1233.
- Savill, J.S.; Wyllie, A.H.; Henson, J.E.; Walport, M.J.; Henson, P.M.; Haslett, C. Macrophage phagocytosis of aging neutrophils in inflammation. Programmed cell death in the neutrophil leads to its recognition by macrophages. J. Clin. Investig. 1989, 83, 865–875.
- Fadok, V.A.; Bratton, D.L.; Konowal, A.; Freed, P.W.; Westcott, J.Y.; Henson, P.M. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J. Clin. Invest. 1998, 101, 890–898.
- Serhan, C.N.; Savill, J. Resolution of inflammation: The beginning programs the end. Nat. Immunol. 2005, 6, 1191–1197.
- Dalli, J.; Serhan, C.N. Pro-Resolving Mediators in Regulating and Conferring Macrophage Function. Front. Immunol. 2017, 8, 1400.
- Mukundan, L.; Odegaard, J.I.; Morel, C.R.; Heredia, J.E.; Mwangi, J.W.; Ricardo-Gonzalez, R.R.; Goh, Y.P.; Eagle, A.R.; Dunn, S.E.; Awakuni, J.U.; et al. PPAR-delta senses and orchestrates clearance of apoptotic cells to promote tolerance. Nat. Med. 2009, 15, 1266–1272.
- Rebe, C.; Raveneau, M.; Chevriaux, A.; Lakomy, D.; Sberna, A.L.; Costa, A.; Bessede, G.; Athias, A.; Steinmetz, E.; Lobaccaro, J.M.; et al. Induction of transglutaminase 2 by a liver X receptor/retinoic acid receptor alpha pathway increases the clearance of apoptotic cells by human macrophages. Circ. Res. 2009, 105, 393–401.
- A-Gonzalez, N.; Bensinger, S.J.; Hong, C.; Beceiro, S.; Bradley, M.N.; Zelcer, N.; Deniz, J.; Ramirez, C.; Diaz, M.; Gallardo, G.; et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity 2009, 31, 245–258.
- Zhang, S.; Weinberg, S.; DeBerge, M.; Gainullina, A.; Schipma, M.; Kinchen, J.M.; Ben-Sahra, I.; Gius, D.R.; Yvan-Charvet, L.; Chandel, N.S.; et al. Efferocytosis Fuels Requirements of Fatty Acid Oxidation and the Electron Transport Chain to Polarize Macrophages for Tissue Repair. Cell Metab. 2019, 29, 443–456 e445.
- Yurdagul, A., Jr.; Subramanian, M.; Wang, X.; Crown, S.B.; Ilkayeva, O.R.; Darville, L.; Kolluru, G.K.; Rymond, C.C.; Gerlach, B.D.; Zheng, Z.; et al. Macrophage Metabolism of Apoptotic Cell-Derived Arginine Promotes Continual Efferocytosis and Resolution of Injury. Cell Metab. 2020, 31, 518–533 e510.
- Hess, C.; Sadallah, S.; Hefti, A.; Landmann, R.; Schifferli, J.A. Ectosomes released by human neutrophils are specialized functional units. J. Immunol. 1999, 163, 4564–4573.
- Gasser, O.; Hess, C.; Miot, S.; Deon, C.; Sanchez, J.C.; Schifferli, J.A. Characterisation and properties of ectosomes released by human polymorphonuclear neutrophils. Exp. Cell Res. 2003, 285, 243–257.
- Dalli, J.; Norling, L.V.; Renshaw, D.; Cooper, D.; Leung, K.Y.; Perretti, M. Annexin 1 mediates the rapid anti-inflammatory effects of neutrophil-derived microparticles. Blood 2008, 112, 2512–2519.
- Szeifert, V.; Kolonics, F.; Bartos, B.; Khamari, D.; Vagi, P.; Barna, L.; Ligeti, E.; Lorincz, A.M. Mac-1 Receptor Clustering Initiates Production of Pro-Inflammatory, Antibacterial Extracellular Vesicles From Neutrophils. Front. Immunol. 2021, 12, 671995.
- Kolonics, F.; Kajdacsi, E.; Farkas, V.J.; Veres, D.S.; Khamari, D.; Kittel, A.; Merchant, M.L.; McLeish, K.R.; Lorincz, A.M.; Ligeti, E. Neutrophils produce proinflammatory or anti-inflammatory extracellular vesicles depending on the environmental conditions. J. Leukoc. Biol. 2021, 109, 793–806.
- Dalli, J.; Serhan, C.N. Specific lipid mediator signatures of human phagocytes: Microparticles stimulate macrophage efferocytosis and pro-resolving mediators. Blood 2012, 120, e60–e72.
- Eken, C.; Sadallah, S.; Martin, P.J.; Treves, S.; Schifferli, J.A. Ectosomes of polymorphonuclear neutrophils activate multiple signaling pathways in macrophages. Immunobiology 2013, 218, 382–392.
- Headland, S.E.; Jones, H.R.; Norling, L.V.; Kim, A.; Souza, P.R.; Corsiero, E.; Gil, C.D.; Nerviani, A.; Dell’Accio, F.; Pitzalis, C.; et al. Neutrophil-derived microvesicles enter cartilage and protect the joint in inflammatory arthritis. Sci. Transl. Med. 2015, 7, 315ra190.
- Rhys, H.I.; Dell’Accio, F.; Pitzalis, C.; Moore, A.; Norling, L.V.; Perretti, M. Neutrophil Microvesicles from Healthy Control and Rheumatoid Arthritis Patients Prevent the Inflammatory Activation of Macrophages. EBioMedicine 2018, 29, 60–69.
- Marwick, J.A.; Mills, R.; Kay, O.; Michail, K.; Stephen, J.; Rossi, A.G.; Dransfield, I.; Hirani, N. Neutrophils induce macrophage anti-inflammatory reprogramming by suppressing NF-kappaB activation. Cell Death Dis. 2018, 9, 665.
- Nauseef, W.M. The phagocyte NOX2 NADPH oxidase in microbial killing and cell signaling. Curr. Opin. Immunol. 2019, 60, 130–140.
- Dinauer, M.C. Primary immune deficiencies with defects in neutrophil function. Hematol. Am. Soc. Hematol. Educ. Program. 2016, 2016, 43–50.
- Coxon, A.; Rieu, P.; Barkalow, F.J.; Askari, S.; Sharpe, A.H.; von Andrian, U.H.; Arnaout, M.A.; Mayadas, T.N. A novel role for the beta 2 integrin CD11b/CD18 in neutrophil apoptosis: A homeostatic mechanism in inflammation. Immunity 1996, 5, 653–666.
- Greenberg, M.E.; Sun, M.; Zhang, R.; Febbraio, M.; Silverstein, R.; Hazen, S.L. Oxidized phosphatidylserine-CD36 interactions play an essential role in macrophage-dependent phagocytosis of apoptotic cells. J. Exp. Med. 2006, 203, 2613–2625.
- Frasch, S.C.; Berry, K.Z.; Fernandez-Boyanapalli, R.; Jin, H.S.; Leslie, C.; Henson, P.M.; Murphy, R.C.; Bratton, D.L. NADPH oxidase-dependent generation of lysophosphatidylserine enhances clearance of activated and dying neutrophils via G2A. J. Biol. Chem. 2008, 283, 33736–33749.
- Frasch, S.C.; Fernandez-Boyanapalli, R.F.; Berry, K.A.; Murphy, R.C.; Leslie, C.C.; Nick, J.A.; Henson, P.M.; Bratton, D.L. Neutrophils regulate tissue Neutrophilia in inflammation via the oxidant-modified lipid lysophosphatidylserine. J. Biol. Chem. 2013, 288, 4583–4593.
- Deng, J.; Wang, X.; Qian, F.; Vogel, S.; Xiao, L.; Ranjan, R.; Park, H.; Karpurapu, M.; Ye, R.D.; Park, G.Y.; et al. Protective role of reactive oxygen species in endotoxin-induced lung inflammation through modulation of IL-10 expression. J. Immunol. 2012, 188, 5734–5740.
- Whitmore, L.C.; Hilkin, B.M.; Goss, K.L.; Wahle, E.M.; Colaizy, T.T.; Boggiatto, P.M.; Varga, S.M.; Miller, F.J.; Moreland, J.G. NOX2 protects against prolonged inflammation, lung injury, and mortality following systemic insults. J. Innate Immun 2013, 5, 565–580.
- Taylor, P.R.; Carugati, A.; Fadok, V.A.; Cook, H.T.; Andrews, M.; Carroll, M.C.; Savill, J.S.; Henson, P.M.; Botto, M.; Walport, M.J. A hierarchical role for classical pathway complement proteins in the clearance of apoptotic cells in vivo. J. Exp. Med. 2000, 192, 359–366.
- Jacob, C.O.; Eisenstein, M.; Dinauer, M.C.; Ming, W.; Liu, Q.; John, S.; Quismorio, F.P., Jr.; Reiff, A.; Myones, B.L.; Kaufman, K.M.; et al. Lupus-associated causal mutation in neutrophil cytosolic factor 2 (NCF2) brings unique insights to the structure and function of NADPH oxidase. Proc. Natl. Acad. Sci. USA 2012, 109, E59–E67.
- Zhong, J.; Olsson, L.M.; Urbonaviciute, V.; Yang, M.; Backdahl, L.; Holmdahl, R. Association of NOX2 subunits genetic variants with autoimmune diseases. Free Radic. Biol. Med. 2018, 125, 72–80.
- Wing, K.; Klocke, K.; Samuelsson, A.; Holmdahl, R. Germ-free mice deficient of reactive oxygen species have increased arthritis susceptibility. Eur. J. Immunol. 2015, 45, 1348–1353.
- Jacob, C.O.; Yu, N.; Yoo, D.G.; Perez-Zapata, L.J.; Barbu, E.A.; Kaplan, M.J.; Purmalek, M.; Pingel, J.T.; Idol, R.A.; Dinauer, M.C. Haploinsufficiency of NADPH Oxidase Subunit Neutrophil Cytosolic Factor 2 Is Sufficient to Accelerate Full-Blown Lupus in NZM 2328 Mice. Arthritis Rheumatol. 2017, 69, 1647–1660.
- Bagaitkar, J.; Huang, J.; Zeng, M.Y.; Pech, N.K.; Monlish, D.A.; Perez-Zapata, L.J.; Miralda, I.; Schuettpelz, L.G.; Dinauer, M.C. NADPH oxidase activation regulates apoptotic neutrophil clearance by murine macrophages. Blood 2018, 131, 2367–2378.
- Brown, J.R.; Goldblatt, D.; Buddle, J.; Morton, L.; Thrasher, A.J. Diminished production of anti-inflammatory mediators during neutrophil apoptosis and macrophage phagocytosis in chronic granulomatous disease (CGD). J. Leukoc. Biol. 2003, 73, 591–599.
- Fernandez-Boyanapalli, R.F.; Frasch, S.C.; McPhillips, K.; Vandivier, R.W.; Harry, B.L.; Riches, D.W.; Henson, P.M.; Bratton, D.L. Impaired apoptotic cell clearance in CGD due to altered macrophage programming is reversed by phosphatidylserine-dependent production of IL-4. Blood 2009, 113, 2047–2055.
- Fernandez-Boyanapalli, R.; Frasch, S.C.; Riches, D.W.; Vandivier, R.W.; Henson, P.M.; Bratton, D.L. PPARgamma activation normalizes resolution of acute sterile inflammation in murine chronic granulomatous disease. Blood 2010, 116, 4512–4522.
- Yang, W.; Tao, Y.; Wu, Y.; Zhao, X.; Ye, W.; Zhao, D.; Fu, L.; Tian, C.; Yang, J.; He, F.; et al. Neutrophils promote the development of reparative macrophages mediated by ROS to orchestrate liver repair. Nat. Commun. 2019, 10, 1076.
More
Information
Subjects:
Cell Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.8K
Revisions:
3 times
(View History)
Update Date:
30 Dec 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No