Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Hannah O'Neill | -- | 2570 | 2022-12-21 01:23:05 | | | |
| 2 | Rita Xu | -12 word(s) | 2558 | 2022-12-21 03:06:07 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
O’neill, H.; Lee, H.; Gupta, I.; Rodger, E.J.; Chatterjee, A. Single-Cell DNA Methylation in Cancer. Encyclopedia. Available online: https://encyclopedia.pub/entry/39008 (accessed on 30 June 2026).
O’neill H, Lee H, Gupta I, Rodger EJ, Chatterjee A. Single-Cell DNA Methylation in Cancer. Encyclopedia. Available at: https://encyclopedia.pub/entry/39008. Accessed June 30, 2026.
O’neill, Hannah, Heather Lee, Ishaan Gupta, Euan J. Rodger, Aniruddha Chatterjee. "Single-Cell DNA Methylation in Cancer" Encyclopedia, https://encyclopedia.pub/entry/39008 (accessed June 30, 2026).
O’neill, H., Lee, H., Gupta, I., Rodger, E.J., & Chatterjee, A. (2022, December 21). Single-Cell DNA Methylation in Cancer. In Encyclopedia. https://encyclopedia.pub/entry/39008
O’neill, Hannah, et al. "Single-Cell DNA Methylation in Cancer." Encyclopedia. Web. 21 December, 2022.
Copy Citation
Cancer is a distinctly difficult disease to treat on account of the diverse cell populations/subpopulations that comprise a tumour. Such cells harbour varying genetic and epigenetic states, which contributes to their oncogenic phenotype. Morphological, transcriptomic, and genomic defects are well-explored parameters of cancer biology. Aberrant DNA methylation has been implicated in many types of cancers, influencing cell type, state, transcriptional regulation, and genomic stability to name a few. Traditionally, large populations of cells from the tissue of interest are coalesced for analysis, producing averaged methylome data.
DNA methylation
single cell
cancer
1. Introduction
Epigenetics has been described as “the study of changes in gene function that are mitotically and/or meiotically heritable and that do not entail a change in DNA sequence” [1]. In essence, the epigenome consists of numerous types of modifications to DNA, which act to regulate the genome [2]. The impact f epigenetics on the cancer genome has been referred to as “the most obvious source of dark matter” [3], exhibiting how elusive the field once was in its contribution to the cancer genome. Epigenetic mechanisms have now been recognised as contributors to the acquisition of cancer hallmark capabilities [4]. Perhaps the most well-explored epigenetic modification, DNA methylation (DNAme), can lead to oncogenic phenotypes when abnormal changes take place [5]. Through transcriptional regulation, altering genomic stability and induction of mutational events to name a few, DNAme heavily influences oncogenic phenotype [6]. Studying such marks and their contribution to oncogenesis is difficult, however, as tumours tend to be highly heterogeneous entities. Cellular methylomes are influenced by cell types and states, of which there are many in a single tumour. Fortunately, recent years have seen the introduction of single-cell sequencing (SCS), a technology that permits the analysis of single-cell methylomes so that comprehensive analyses of epigenetic heterogeneity are now possible.
1.1. DNA Methylation
Epigenetic modifications are described as chemical modifications to the DNA that cause a heritable phenotype without changing the primary DNA sequence [7]. One of the main epigenetic alterations is DNA methylation (DNAme), a stable yet reversible mark entailing the addition of a methyl group, typically at the fifth position of a cytosine residue (5-methylcytosine, 5mC) adjacent to a guanine residue (CpG site) [8]. A total of 28.7 million CpG sites are present in the human genome; these CpGs are often found concentrated together, forming CpG islands (CGIs), which are regions of the genome with a higher frequency of CpGs than expected [9]. Usually, these CGIs are present in promoter regions of genes, and the presence or lack of DNA methylation influences the transcriptional activity of the associated gene. In most cases, the presence of DNA methylation (DNAme) at CGIs represses transcription, whereas DNAme absence is observed in transcriptionally active genes [10]. These regulatory properties of DNAme are crucial in numerous fundamental biological processes throughout the lifespan, including cell-cycle control, cell fate decisions, X-chromosome inactivation, genomic imprinting, embryonic development, chromosomal stability, and transposable element silencing [11][12][13][14]. Considering the above, it is not surprising that aberrant DNAme patterns are implicated in many diseases, including Alzheimer’s [15], cardiovascular diseases [16], all types of cancer [17]. Furthermore, aberrations in enzymes associated with DNAme, such as ten-eleven translocation proteins, have been identified as cancer hallmarks [18], as well as intermediate states of 5mC, such as 5-hydroxymethylcytosine (5hmC) [19][20].
1.2. Single-Cell DNA Methylation
Traditionally, due to technical limitations, large numbers of cells have been combined and analysed as one to obtain insight into DNAme patterns. For conciseness, this practice will be referred to as “bulk sequencing”. When combining thousands of cells for analysis, an averaged methylome profile of all the cells will be produced. While this technique has been excellent in advancing the knowledge of the role DNAme plays in disease, there are some areas of research that bulk sequencing is not capable of reaching. For example, small subpopulations with distinct methylomes within tumours are present with greater metastatic capabilities; however, their methylomes will be masked by the many other cells present in the tumour [21].
Recent years have seen the emergence of single-cell technologies, allowing for the analysis of single cells’ epigenomes (Figure 1). In essence, this means cells with differing methylomes reflecting different cell types/states in a tumour can now be identified and appreciated as differing components of the tumour, rather than grouped together as one. This allows for analyses, such as characterisation of rare subpopulations and cell lineage tracing, which provide insight into the epigenetic regulation of heterogeneous populations of cells.
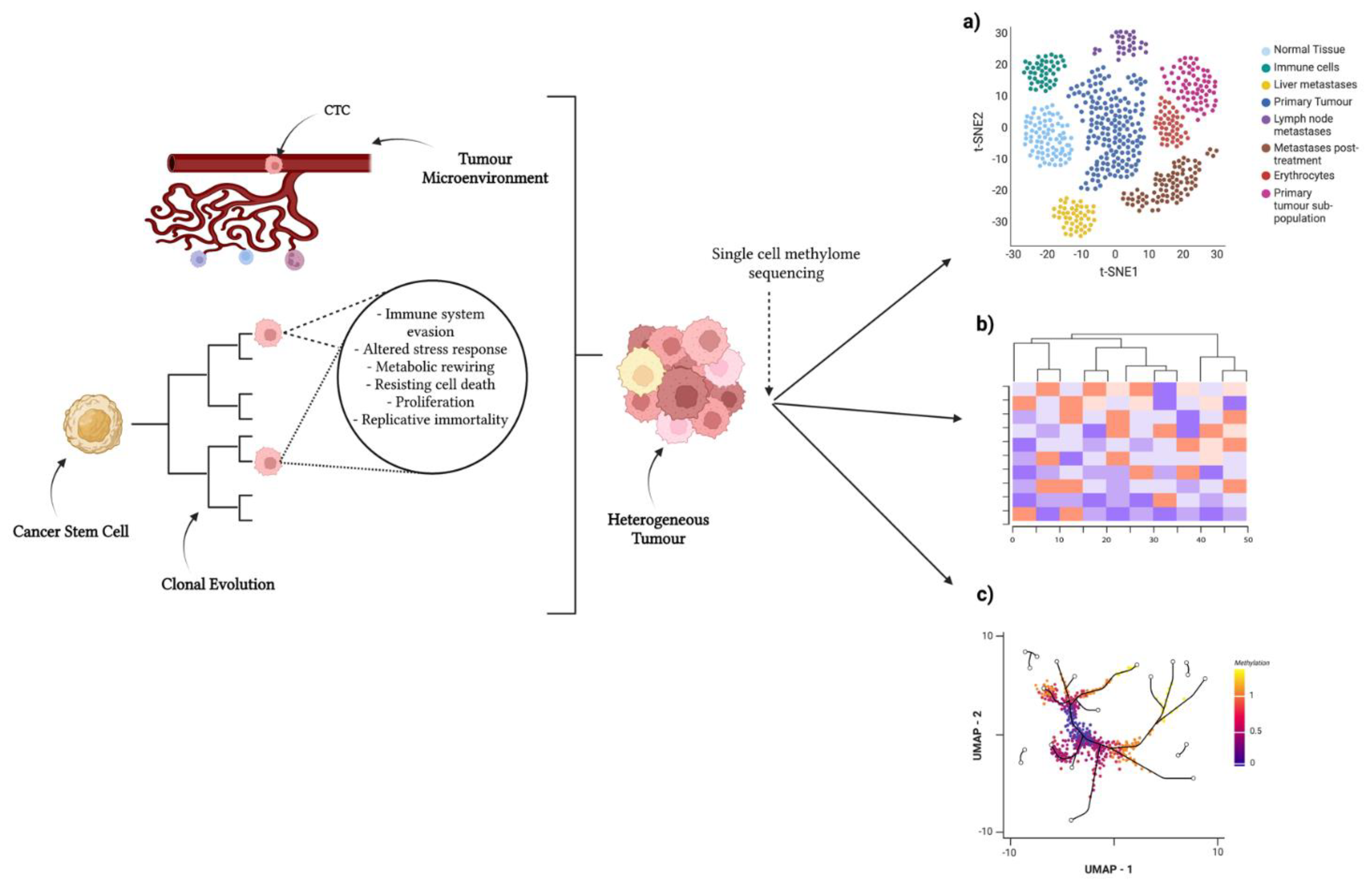
Figure 1. Single-cell approaches to identify heterogeneity in tumour population. Tumour-initiating stem cells develop in tissue of origin. These undergo branched tumour evolution, acquiring random mutations and epigenomic alterations. The tumour microenvironment influences heterogeneity via physical and chemical signals. A combinatorial effect of these concepts induces a highly heterogeneous tumour, as well as circulating tumour cells (CTCs) disseminating from said tumour. Single-cell methylome sequencing of tumours can provide insight into subpopulations/differing cell states by clustering cells: (a) clustering via t-SNE, (b) clustering via UMAP, and (c) cell lineages/differentially methylated regions via heatmaps. Created with https://biorender.com/ (accessed on 25 July 2022).
2. Single-Cell Methylome Profiling in Cancer
Collectively, the genome, epigenome, and transcriptome of cancer cells are grossly dysregulated. The exploitation of pathways required in normal biological processes results in the initiation, progression, dissemination, and metastasis of cancer. As a result of inactive tumour suppressor genes or overactive oncogenes, processes such as apoptosis, cell proliferation, and DNA repair mechanisms are interrupted [22], while others, such as angiogenesis, replicative immortality, and the ability to evade growth suppression, are influenced [23]. Epigenetic modifications have been implicated in cancer predisposition, early tumorigenesis events, metastasis, and therapeutic resistance [24].
A recurrent challenge faced while studying most types of cancer is its heterogenic nature. Heterogeneity refers to the state of being diverse. In the context of cancer, this can reflect the diversity between individuals (inter-individual heterogeneity), between primary tumours and their metastases (inter-tumoural), and between cells within the same tumour (intra-tumoural). Tumours are comprised of numerous types of cells, including the malignant and healthy cells from the tissue of origin, fibroblasts, immune cells, nerves, and subpopulations of tumour cells [25]. Single-cell sequencing provides a tool for understanding the complex dynamics and heterogeneity that encompasses each tumour.
2.1. Single-Cell DNA Methylation in Cancer Initiation and Progression
The diversification of cells begins with cancer stem cells from the tissue of origin. These cells have self-renewal capabilities and multi-lineage differentiation, which encourage tumorigenesis and heterogeneity [26]. Cancer stem cells are known to drive initiation and progression, as well as encourage disease relapse [27]. Famously, cancer stem cells have been difficult to isolate due to their similarities to normal tissue-of-origin stem cells and their scarcity. The emergence of SCS means it is now possible to isolate cancer stem cells and study them to a higher resolution, without noise from other cells’ epigenomes influencing the data. One study was able to identify a small number of tumour-initiating cells in human glioma through the use of SCS [28], albeit with single-cell RNA sequencing; however, the same principles could be achieved with single-cell methylome techniques. This group was able to evidence that glioblastoma development occurs along with conserved neurodevelopmental gene processes, as well as potential sources of resistance to some immunotherapies in these cancer stem cells.
Tumour cells heavily influence their surrounding environment, inducing chemical, molecular and physical changes. The hallmark features of the tumour microenvironment include blood vessels, immune cells, stromal cells, and extracellular matrix [29]. Following Darwinian processes, tumour cells are continuously interacting and being influenced by the tumour microenvironment, driving branched tumour evolution of cells with selectively advantageous traits to survive [30]. Branched tumour evolution describes when distinct subclones within the tumour evolve in parallel, leading to a considerable amount of diversity between subclones [31]. Phenotypically, this evolution often contributes to evading the immune system, angiogenesis, and avoiding cellular growth-suppressing processes, encouraging tumour progression [32]. In consequence, a highly heterogeneous tumour is formed. Therefore, when combining a large population of tumour cells for analysis, the true individual cellular methylomes are averaged by neighbouring cells. As such, it is expected that cells within a tumour will exhibit great variation, encouraging the need for higher resolution techniques, such as single cell. One group utilised single-cell DNA methylome data to identify the lineage tree of subpopulations in human chronic lymphocytic leukaemia [33]. The lineage tree was created based on the epigenetic aberrations of the cells, showing different chronic lymphocytic leukaemia lineages that were discriminatively influenced and expelled from lymph nodes by ibrutinib, one of the treatments for chronic lymphocytic leukaemia.
The differentiation of cancer stem cells, followed by interactions with the tumour microenvironment driving branched tumour evolution are key events that produce the heterogeneity observed within and between tumours. The use of SCS technologies will not only augment the current understanding of these events, but possibly introduce new pathways for investigation as to how tumours initiate and progress.
2.2. Single-Cell DNA Methylation in Metastasis
Cancer dissemination and subsequent metastasis to new niches around the body is one of the leading causes of death in cancer patients [34]. Circulating tumour cells (CTCs) are highly tumorigenic cells that can lead to metastasis and, due to the low number of CTCs in patient blood, the epigenetic landscape of CTCs remains less characterised [35]. Previously, the low DNA content meant bulk sequencing was not an option for the analysis of CTCs. Using SCS to analyse CTCs provides opportunity to investigate the epigenetic processes that may drive metastatic dissemination of cancer cells and subsequent invasions at new locations around the body. SCS was used to show that CTCs from breast cancer patients showed a focal hypomethylation at a number of stem cell gene promoters, increasing the expression of pluripotency networks [36].
As well as studying CTCs, being able to isolate and analyse subclonal populations which have evolved from the primary to metastatic tumour sites may give insight into the pathways which are exploited for this process. One study used SCS on two secondary tumours (metastatic breast and metastatic castration-resistant prostate cancer), examining methylation patterns on epithelial–mesenchymal transition (EMT)-associated genes to predict metastatic potential [37]. This identified differentially methylated patterns between the two entities, showing that the miR200 feedback loops, involved in inducing epithelial differentiation in the EMT process, were differentially regulated. Another group reconstructed the lineage within a colorectal cancer patient between the primary tumour and its associated metastases and found global DNAme was stable during metastasis, with no DNAme alterations in EMT-related genes prior to or post metastasis [38]. This group also showed a sub-lineage within the primary tumour was detected in the lymph node and liver metastases of the patient, suggesting these metastases had a common origin.
2.3. Single-Cell DNA Methylation in Cancer Therapy
The role DNAme plays in relation to cancer initiation and progression has made DNAme and its regulators a well-explored target for cancer therapy [39]. Furthermore, studies have been able to predict responses to therapy through the use of DNAme biomarkers. For instance, glioblastoma patients with methylated MGMT promoters responded better to alkylating drugs than individuals without methylation [19]. Different subpopulations may have these methylation alterations at these important loci as a result of the branched tumour evolution outlined earlier, and SCS has the ability to uncover such populations. Furthermore, aberrant DNAme accumulation in cancer stem cells was shown to affect self-renewal capabilities, differentiation, multi-drug resistance, and metastasis processes in cancer stem cells [40]. As previously mentioned, cancer stem cells make up a minor population of the tumour, with far greater capabilities of responding to radio- and chemotherapies [26]. SCS could, therefore, enable the detection of cancer stem cells, as well as the discovery of novel biomarkers for therapeutically resistant cells. SCS has been used in a number of papers to look into issues that are clinically significant. For instance, it was utilised to compare the tumour microenvironment before and after immunotherapy in basal cell carcinoma [41]. This provided insight into the regulatory networks controlling cellular response to therapy, in particular, some overlapping regulation of therapy-responsive T cells. Moreover, different epigenomic subpopulations have been shown to vary in their response to targeted therapy, with certain subpopulations showing greater resistance to imatinib [42]. Understanding the molecular heterogeneity of the individual’s tumour could also be used to inform oncologists on which currently available treatments could be combined for the most effective treatment.
In a clinical context, it has also been proposed that SCS could provide noninvasive tests by taking liquid biopsies containing CTCs at different time points throughout a patient’s therapy [43]. Multiple studies have found that CTCs had over 50% of the same mutations as the primary tumour in lung cancer [44] and colon cancer [45]. Therefore, they could be extracted from blood and analysed to track the evolution of the primary tumour and adjust treatment accordingly. Another study found there was a substantial amount of methylome heterogeneity amongst distinct CTCs from the same patients [37]. This could lead to personalised signatures in these CTCs for patient stratification and treatment selection. Circulating tumour DNA (ctDNA) is tumour-derived DNA that has become free in the blood [46]. While ctDNA has also been used to extract epigenetic signatures from liquid biopsies for similar purposes to CTCs, CTCs represent still intact tumour cells, which are likely to give more comprehensive information regarding the tumour it disseminated from [47].
Single-cell techniques are most commonly used for RNA analyses to profile gene expression rather than DNAme analyses. A typical cell contains approximately 10–30 pg of RNA, which is then reverse transcribed into cDNA and amplified [48]. Conversely, the starting material for single-cell DNAme analyses starting material is only the double-stranded DNA (~6 pg maximum) [49]. The native DNA then requires manipulation, such as bisulphite conversion or enzymatic treatment prior to amplification; therefore, there is a high risk of DNA loss [50]. As such, the disparity between the number of publications of the two techniques can likely be attributed to the significantly lower starting material of DNAme and the library preparation techniques required and technical challenges associated with single-cell DNAme experiments (Table 1). Nonetheless, the epigenetic influence on oncogenic phenotypes and cancers’ inherent heterogeneity is a well-appreciated phenomenon, which warrants further investigation in the single-cell context. Moreover, DNAme is a stable epigenetic mark that is likely to represent a sustained signature in contrast to gene expression changes, which are more variable and represent dynamic changes in the cell. With the above points considered, single-cell DNAme sequencing is an enticing direction for the field, despite the challenges currently being faced.
Table 1. Examples of single-cell methylome sequencing experiments in cancer.
| Cancer Type | Genome-Wide or Gene-Specific | Findings | Year Published | References |
|---|---|---|---|---|
| HL60 (acute promyelocytic leukemia cell line) and K562 (erythroleukemia-derived cell line) | Genome-wide | First implementation of single-cell WGBS | 2015 | [51] |
| Hepatocellular carcinoma | Genome-wide | Identification of subpopulations within tumour | 2016 | [52] |
| Metastatic breast cancer (mBC) and metastatic castration-resistant prostate cancer (mCRPC) |
CDH1 and miR200 promoters. | CTCs from same patient displayed heterogeneous methylation patterns. Different methylation patterns at these promoters in mCRPC vs. mBC CTCs suggesting differentially regulated miR200 loops in these two tumour entities. | 2017 | [37] |
| Colorectal cancer | Genome-wide | Sub-lineages identified in patients found metastases at multiple sites had a common origin | 2018 | [38] |
| Chronic Lymphocytic Leukaemia | Genome-wide | Subpopulations preferentially expelled from lymph nodes after treatment | 2019 | [33] |
| Lung Adenocarcinoma | Genome-wide | Global methylation heterogeneity amongst tumours associated with the progression of LAC | 2021 | [53] |
| Lung Cancer | Genome-wide | Unique CTC DNA methylation signature distinguished it from the primary tumour | 2021 | [54] |
| 6 Cancer Types: Prostate, Colon, Small cell lung, Lung Adenocarcinoma, Breast, and Gastric | Genome-wide | Potential to identify tumours of origin for CTC based on methylome profiles. Report diverse evolutionary histories of CTCs | 2021 | [47] |
| KG1a Acute Myeloid Leukaemia | Transposable elements: SINE Alu | TEs as a surrogate for predicting single-cell global DNA methylation. Method has greater alignment and costs 3-fold less than scBS-seq | 2022 | [55] |
References
- Wu, C.; Morris, J.R. Genes, genetics, and epigenetics: A correspondence. Science 2001, 293, 1103–1105.
- Dupont, C.; Armant, D.R.; Brenner, C.A. Epigenetics: Definition, mechanisms and clinical perspective. Semin. Reprod. Med. 2009, 27, 351–357.
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558.
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46.
- Chatterjee, A.; Rodger, E.J.; Eccles, M.R. Epigenetic drivers of tumourigenesis and cancer metastasis. Semin. Cancer Biol. 2018, 51, 149–159.
- Banerjee, R.; Smith, J.; Eccles, M.R.; Weeks, R.J.; Chatterjee, A. Epigenetic basis and targeting of cancer metastasis. Trends Cancer 2022, 8, 226–241.
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. Circulation 2011, 123, 2145–2156.
- Jin, B.; Li, Y.; Robertson, K.D. DNA methylation: Superior or subordinate in the epigenetic hierarchy? Genes Cancer 2011, 2, 607–617.
- Kulis, M.; Esteller, M. 2—DNA Methylation and Cancer. In Advances in Genetics; Herceg, Z., Ushijima, T., Eds.; Academic Press: Cambridge, MA, USA, 2010; Volume 70, pp. 27–56.
- Wade, P.A. Methyl CpG-binding proteins and transcriptional repression. Bioessays 2001, 23, 1131–1137.
- Seisenberger, S.; Andrews, S.; Krueger, F.; Arand, J.; Walter, J.; Santos, F.; Popp, C.; Thienpont, B.; Dean, W.; Reik, W. The dynamics of genome-wide DNA methylation reprogramming in mouse primordial germ cells. Mol. Cell 2012, 48, 849–862.
- Walsh, C.P.; Chaillet, J.R.; Bestor, T.H. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat. Genet. 1998, 20, 116–117.
- Bartolomei, M.S. Genomic imprinting: Employing and avoiding epigenetic processes. Genes Dev. 2009, 23, 2124–2133.
- Hellman, A.; Chess, A. Gene body-specific methylation on the active X chromosome. Science 2007, 315, 1141–1143.
- Shireby, G.; Dempster, E.L.; Policicchio, S.; Smith, R.G.; Pishva, E.; Chioza, B.; Davies, J.P.; Burrage, J.; Lunnon, K.; Seiler Vellame, D.; et al. DNA methylation signatures of Alzheimer’s disease neuropathology in the cortex are primarily driven by variation in non-neuronal cell-types. Nat. Commun. 2022, 13, 5620.
- Fernández-Sanlés, A.; Sayols-Baixeras, S.; Subirana, I.; Sentí, M.; Pérez-Fernández, S.; de Castro Moura, M.; Esteller, M.; Marrugat, J.; Elosua, R. DNA methylation biomarkers of myocardial infarction and cardiovascular disease. Clin. Epigenet. 2021, 13, 86.
- Kandi, V.; Vadakedath, S. Effect of DNA Methylation in Various Diseases and the Probable Protective Role of Nutrition: A Mini-Review. Cureus 2015, 7, e309.
- Yang, H.; Liu, Y.; Bai, F.; Zhang, J.Y.; Ma, S.H.; Liu, J.; Xu, Z.D.; Zhu, H.G.; Ling, Z.Q.; Ye, D.; et al. Tumor development is associated with decrease of TET gene expression and 5-methylcytosine hydroxylation. Oncogene 2013, 32, 663–669.
- Haffner, M.C.; Chaux, A.; Meeker, A.K.; Esopi, D.M.; Gerber, J.; Pellakuru, L.G.; Toubaji, A.; Argani, P.; Iacobuzio-Donahue, C.; Nelson, W.G.; et al. Global 5-hydroxymethylcytosine content is significantly reduced in tissue stem/progenitor cell compartments and in human cancers. Oncotarget 2011, 2, 627–637.
- Rodger, E.J.; Chatterjee, A.; Morison, I.M. 5-hydroxymethylcytosine: A potential therapeutic target in cancer. Epigenomics 2014, 6, 503–514.
- Nguyen, A.; Yoshida, M.; Goodarzi, H.; Tavazoie, S.F. Highly variable cancer subpopulations that exhibit enhanced transcriptome variability and metastatic fitness. Nat. Commun. 2016, 7, 11246.
- Kastan, M.B.; Canman, C.E.; Leonard, C.J. P53, cell cycle control and apoptosis: Implications for cancer. Cancer Metastasis Rev. 1995, 14, 3–15.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Pan, Y.; Liu, G.; Zhou, F.; Su, B.; Li, Y. DNA methylation profiles in cancer diagnosis and therapeutics. Clin. Exp. Med. 2018, 18, 1–14.
- Wu, F.; Fan, J.; He, Y.; Xiong, A.; Yu, J.; Li, Y.; Zhang, Y.; Zhao, W.; Zhou, F.; Li, W.; et al. Single-cell profiling of tumor heterogeneity and the microenvironment in advanced non-small cell lung cancer. Nat. Commun. 2021, 12, 2540.
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. J. Biomed. Sci. 2018, 25, 20.
- Walcher, L.; Kistenmacher, A.-K.; Suo, H.; Kitte, R.; Dluczek, S.; Strauß, A.; Blaudszun, A.-R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer Stem Cells—Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front. Immunol. 2020, 11, 1280.
- Couturier, C.P.; Ayyadhury, S.; Le, P.U.; Nadaf, J.; Monlong, J.; Riva, G.; Allache, R.; Baig, S.; Yan, X.; Bourgey, M.; et al. Single-cell RNA-seq reveals that glioblastoma recapitulates a normal neurodevelopmental hierarchy. Nat. Commun. 2020, 11, 3406.
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925.
- Álvarez-Arenas, A.; Podolski-Renic, A.; Belmonte-Beitia, J.; Pesic, M.; Calvo, G.F. Interplay of Darwinian Selection, Lamarckian Induction and Microvesicle Transfer on Drug Resistance in Cancer. Sci. Rep. 2019, 9, 9332.
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313.
- Greaves, M. Cancer: The Evolutionary Legacy; Oxford University Press on Demand: Oxford, UK, 2001.
- Gaiti, F.; Chaligne, R.; Gu, H.; Brand, R.M.; Kothen-Hill, S.; Schulman, R.C.; Grigorev, K.; Risso, D.; Kim, K.T.; Pastore, A.; et al. Epigenetic evolution and lineage histories of chronic lymphocytic leukaemia. Nature 2019, 569, 576–580.
- Dillekås, H.; Rogers, M.S.; Straume, O. Are 90% of deaths from cancer caused by metastases? Cancer Med. 2019, 8, 5574–5576.
- Vasantharajan, S.S.; Eccles, M.R.; Rodger, E.J.; Pattison, S.; McCall, J.L.; Gray, E.S.; Calapre, L.; Chatterjee, A. The Epigenetic landscape of Circulating tumour cells. Biochim. Biophys. Acta (BBA) Rev. Cancer 2021, 1875, 188514.
- Gkountela, S.; Castro-Giner, F.; Szczerba, B.M.; Vetter, M.; Landin, J.; Scherrer, R.; Krol, I.; Scheidmann, M.C.; Beisel, C.; Stirnimann, C.U.; et al. Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding. Cell 2019, 176, 98–112.e114.
- Pixberg, C.F.; Raba, K.; Müller, F.; Behrens, B.; Honisch, E.; Niederacher, D.; Neubauer, H.; Fehm, T.; Goering, W.; Schulz, W.A.; et al. Analysis of DNA methylation in single circulating tumor cells. Oncogene 2017, 36, 3223–3231.
- Bian, S.; Hou, Y.; Zhou, X.; Li, X.; Yong, J.; Wang, Y.; Wang, W.; Yan, J.; Hu, B.; Guo, H.; et al. Single-cell multiomics sequencing and analyses of human colorectal cancer. Science 2018, 362, 1060–1063.
- Seymour, J.F.; Döhner, H.; Butrym, A.; Wierzbowska, A.; Selleslag, D.; Jang, J.H.; Kumar, R.; Cavenagh, J.; Schuh, A.C.; Candoni, A.; et al. Azacitidine improves clinical outcomes in older patients with acute myeloid leukaemia with myelodysplasia-related changes compared with conventional care regimens. BMC Cancer 2017, 17, 852.
- Mazloumi, Z.; Farahzadi, R.; Rafat, A.; Dizaji Asl, K.; Karimipour, M.; Montazer, M.; Movassaghpour, A.A.; Dehnad, A.; Nozad Charoudeh, H. Effect of aberrant DNA methylation on cancer stem cell properties. Exp. Mol. Pathol. 2022, 125, 104757.
- Satpathy, A.T.; Granja, J.M.; Yost, K.E.; Qi, Y.; Meschi, F.; McDermott, G.P.; Olsen, B.N.; Mumbach, M.R.; Pierce, S.E.; Corces, M.R.; et al. Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat. Biotechnol. 2019, 37, 925–936.
- Litzenburger, U.M.; Buenrostro, J.D.; Wu, B.; Shen, Y.; Sheffield, N.C.; Kathiria, A.; Greenleaf, W.J.; Chang, H.Y. Single-cell epigenomic variability reveals functional cancer heterogeneity. Genome Biol. 2017, 18, 15.
- Wang, Y.; Navin, N.E. Advances and applications of single-cell sequencing technologies. Mol. Cell 2015, 58, 598–609.
- Ni, X.; Zhuo, M.; Su, Z.; Duan, J.; Gao, Y.; Wang, Z.; Zong, C.; Bai, H.; Chapman, A.R.; Zhao, J.; et al. Reproducible copy number variation patterns among single circulating tumor cells of lung cancer patients. Proc. Natl. Acad. Sci. USA 2013, 110, 21083–21088.
- Lohr, J.G.; Adalsteinsson, V.A.; Cibulskis, K.; Choudhury, A.D.; Rosenberg, M.; Cruz-Gordillo, P.; Francis, J.M.; Zhang, C.Z.; Shalek, A.K.; Satija, R.; et al. Whole-exome sequencing of circulating tumor cells provides a window into metastatic prostate cancer. Nat. Biotechnol. 2014, 32, 479–484.
- Tan, C.R.C.; Zhou, L.; El-Deiry, W.S. Circulating Tumor Cells Versus Circulating Tumor DNA in Colorectal Cancer: Pros and Cons. Curr. Color. Cancer Rep. 2016, 12, 151–161.
- Chen, H.; Su, Z.; Li, R.; Zhang, N.; Guo, H.; Bai, F. Single-cell DNA methylome analysis of circulating tumor cells. Chin. J. Cancer Res. 2021, 33, 391–404.
- Huang, X.T.; Li, X.; Qin, P.Z.; Zhu, Y.; Xu, S.N.; Chen, J.P. Technical Advances in Single-Cell RNA Sequencing and Applications in Normal and Malignant Hematopoiesis. Front. Oncol. 2018, 8, 582.
- Russo, J.; Sheriff, F.; Cicco, R.L.D.; Pogash, T.J.; Nguyen, T.; Russo, I.H. Chapter 3-Methodology for Studying the Compartments of the Human Breast; Springer: New York, NY, USA, 2014; p. 95.
- Ahn, J.; Heo, S.; Lee, J.; Bang, D. Introduction to Single-Cell DNA Methylation Profiling Methods. Biomolecules 2021, 11, 1013.
- Farlik, M.; Sheffield, N.C.; Nuzzo, A.; Datlinger, P.; Schönegger, A.; Klughammer, J.; Bock, C. Single-cell DNA methylome sequencing and bioinformatic inference of epigenomic cell-state dynamics. Cell Rep. 2015, 10, 1386–1397.
- Hou, Y.; Guo, H.; Cao, C.; Li, X.; Hu, B.; Zhu, P.; Wu, X.; Wen, L.; Tang, F.; Huang, Y.; et al. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res. 2016, 26, 304–319.
- Chen, Q.-F.; Gao, H.; Pan, Q.-Y.; Wang, Y.-J.; Zhong, X.-N. Analysis at the single-cell level indicates an important role of heterogeneous global DNA methylation status on the progression of lung adenocarcinoma. Sci. Rep. 2021, 11, 23337.
- Zhao, L.; Wu, X.; Zheng, J.; Dong, D. DNA methylome profiling of circulating tumor cells in lung cancer at single base-pair resolution. Oncogene 2021, 40, 1884–1895.
- Hunt, K.V.; Burnard, S.M.; Roper, E.A.; Bond, D.R.; Dun, M.D.; Verrills, N.M.; Enjeti, A.K.; Lee, H.J. scTEM-seq: Single-cell analysis of transposable element methylation to link global epigenetic heterogeneity with transcriptional programs. Sci. Rep. 2022, 12, 5776.
More
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
916
Revisions:
2 times
(View History)
Update Date:
21 Dec 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No