Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Wenxue Ma | -- | 4854 | 2022-12-15 02:42:05 | | | |
| 2 | Beatrix Zheng | Meta information modification | 4854 | 2022-12-16 03:41:02 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Chen, Q.; Lu, L.; Ma, W. CAR T-Cells in the Treatment of Solid Tumors. Encyclopedia. Available online: https://encyclopedia.pub/entry/38792 (accessed on 25 June 2026).
Chen Q, Lu L, Ma W. CAR T-Cells in the Treatment of Solid Tumors. Encyclopedia. Available at: https://encyclopedia.pub/entry/38792. Accessed June 25, 2026.
Chen, Qiuqiang, Lingeng Lu, Wenxue Ma. "CAR T-Cells in the Treatment of Solid Tumors" Encyclopedia, https://encyclopedia.pub/entry/38792 (accessed June 25, 2026).
Chen, Q., Lu, L., & Ma, W. (2022, December 15). CAR T-Cells in the Treatment of Solid Tumors. In Encyclopedia. https://encyclopedia.pub/entry/38792
Chen, Qiuqiang, et al. "CAR T-Cells in the Treatment of Solid Tumors." Encyclopedia. Web. 15 December, 2022.
Copy Citation
Chimeric antigen receptor (CAR) T-cell therapy is a newly designed adoptive immunotherapy that is able to target and further eliminate cancer cells by engaging with MHC-independent tumor-antigens. CAR T-cell therapy has exhibited conspicuous clinical efficacy in hematological malignancies, but more than half of patients will relapse.
CAR (chimeric antigen receptor)
antigen
heterogeneity
efficacy
safety
T cell exhaustion
1. Introduction
According to the American Cancer Society (www.cancer.org (accessed on 18 November 2022)), cancer remains the second most common cause of deaths in the United States, just behind heart disease. As of 2022, it is estimated that there have been 1.9 million new cancer cases and 609,360 deaths in the United States, or about 1669 deaths every day [1].
In addition to the traditional cancer treatments, such as surgery, chemotherapy, radiation therapy, and targeted therapies (e.g., Imatinib/Gleevec, Trastuzumab/Herceptin, etc.), immunotherapy, which re-activates immune defenses against cancer cells, is a promising new approach and has emerged as the fifth pillar of cancer treatment in the last decade.
Cell-based immunotherapy is effective in cancers. Activated T cells recognize tumor antigens in the form of tumor antigenic peptide fragments that are presented and bound and bind to major histocompatibility complex class (MHC) molecules on the surface of tumor cells. However, the failure of T cells to eradicate cancer cells can result from the expression of immune checkpoint proteins on the surface of T cells, including different mechanisms such as T-cell exhaustion and immunosuppression [2]. Over the past decade, a number of immune checkpoint molecules including programmed cell death protein 1 (PD-1), cytotoxic T lymphocyte antigen-4 (CTLA-4), lymphocyte-activation gene 3 (LAG3), T cell immunoglobulin and mucin domain-containing protein 3 (TIM3), T cell immunoreceptor with Ig and ITIM domains (TIGIT), and B- and T-lymphocyte attenuator (BTLA, also known as CD272) have been identified and well-studied in cancer [2][3].
One of the upmost achievements in cancer immunotherapies in the last decade has undoubtedly been the introduction of T cell-targeted immunomodulators, the immune checkpoint inhibitors (ICIs), such as antibodies against CTLA-4 and PD-1 or programmed death ligand-1 (PD-L1) [4]. The first ICI antibody, Ipilimumab (Yervoy), was approved in 2011, followed by Pembrolizumab (Keytruda) and Nivolumab (Opdivo) in 2014. These ICIs have been widely used to treat a variety of cancer types, including head and neck, lung, kidney, bladder, lymphoma, and melanoma, etc. [2]. These ICI-based immunotherapies are not focused on in this research.
Chimeric antigen receptor (CAR) T cell (CAR T-cell)-based immunotherapy is another greatest achievement in cancer immunotherapies. CARs are generated by artificially fusing a tumor-specific antibody single-chain variable fragment (scFv) to the CD3ζ chain of the T cell receptor (TCR) via a transmembrane linker domain. The scFv specifically recognize specific antigens expressed on the cancer cell surface, or intracellular antigens if the scFv is expressed as intracellular antibody (intrabody) or delivered into the cells [5]. Thus, CAR is a combination of antibody-derived extracellular proteins, typically derived from the intracellular signaling module of an antibody and T-cell signaling proteins [6]. The fusion constructs are then transfected into autologous or allogeneic cytolytic T lymphocytes as CAR T-cells.
CAR T-cells recognize and target tumor antigens through the binding of CAR to tumor-associated antigen (TAA) or tumor-specific antigen (TSA) independent of the TCR-MHC/peptide interaction [6]. CAR T-cells have emerged as an effective novel cancer therapy for hematological malignancies [7].
The flow of the production, application and monitoring of CAR T-cells is summarized in Figure 1. CAR T-cell activity can be monitored with flow cytometric assay [8]. In clinical application, lymphodepletion is needed before the infusion of CAR T-cells to patients, so that the persistence of infused CAR T-cells can be effectively prolonged, and the effectiveness of tumor treatment can be improved [9].
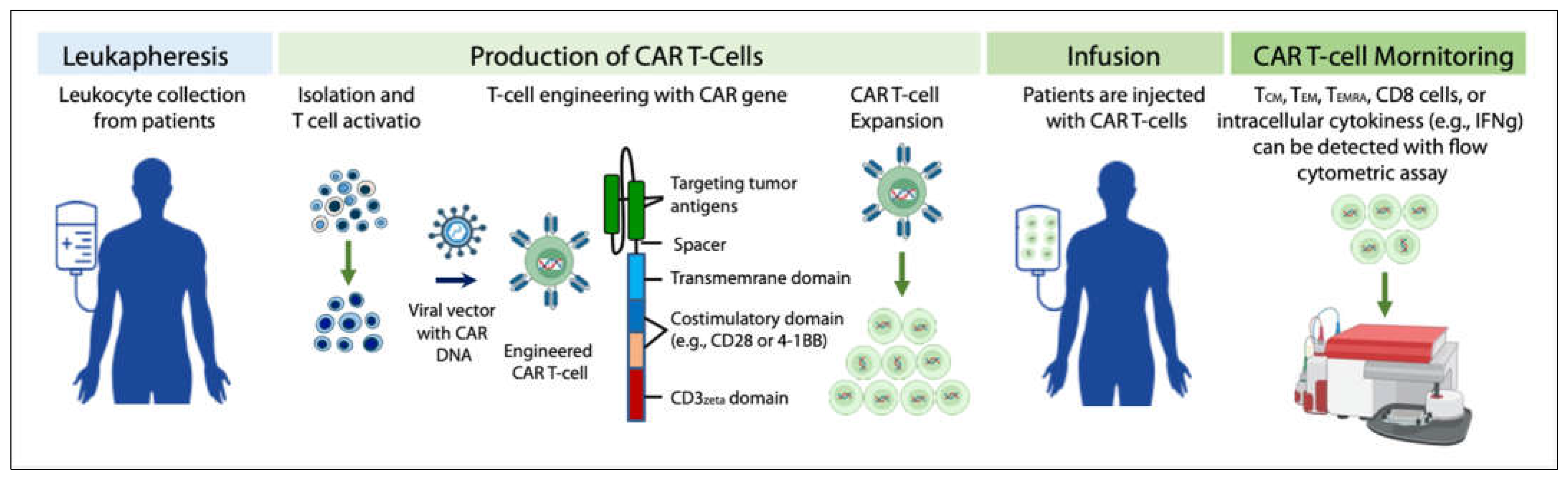
Figure 1. Production, application, and activity monitoring of CAR T-cells.
The specificity of T cells against tumor cells is mediated by CAR proteins. At present, pan-B cell CD19 CAR T-cells have shown unprecedented response rates in treating hematological malignancies including refractory (R/R) B cell malignancies [10][11][12]. In 2017, autologous anti-CD19 CAR T-cells received the first regulatory approval from the US Food and Drug Administration (FDA) for the treatment of pediatric B-cell acute lymphoblastic leukemia (B-ALL), diffuse large B cell lymphoma (DLBCL), and, more recently, mantle cell lymphoma (MCL) [13].
T cells isolated from a cancer patient (for making autologous CAR T-cells), or other healthy donors (for making allogeneic CAR T-cells) are activated using artificial antigen-presenting cells (aAPCs), transfected with the CAR-encoding viral vector, and then expanded to large numbers in a bioreactor. After expansion, the cells are washed followed by the infusion back to the patient or concentrated and cryopreserved for future use. Activity of circulating CAR T-cells can be monitored with flow cytometry at different time points (e.g., 1 month, 3 months, 6 months, etc.) [14] by staining the antibodies of CD62L (circulating innate lymphoid cell precursors) [15], CD45RO (memory T cells), CD45RA (naïve T cells), CD4 (T helper cells), and CD8 (cytotoxic T lymphocytes). Subpopulations were defined as CD62L+ CD45RO+ central memory T cells (TCM), CD62L- CD45RO+ effector memory T cells (TEM), CD62L- CD45RA+ cells (TEMRA), and CD62L+ CD45RA+ naïve T cells [8]. Cytokines including intracellular IFN-γ etc. can also be measured by flow cytometry [16].
A decade ago, CD19-targeting CAR T-cells showed efficacy in patients with chronic lymphocytic leukemia (CLL) [17] and ALL [18]. However, the wide clinical application of CAR T-cells in both diseases has stalled. Patients with CLL did not respond to CD19 CAR T-cells as often as expected, at least in part because of challenges of producing CAR T-cell using autologous T cells from the patients with underlying diseases, or long term chemotherapy treatment, which can lead to lymphocytopenia [19]. Meanwhile, severe lymphocytopenia caused by chemotherapy is associated with reduced survival [20][21]. It is important to note that lymphocytopenia is a different concept from lymphodepletion. Lymphocytopenia is a disorder that lacks lymphocytes. In contrast, lymphodepletion is to purposely eradicate regulatory T-cells (Tregs) and other immunosuppressive cells through treatments and to make a room for the new CAR T-cells, thereby increasing CAR T-cell expansion prolonging persistence [21][22][23].
Due to the impressive results of CAR T-cells in hematological malignancies, many scientists in academia and industries have been attempting to extend CAR T- therapy to solid tumors. In recent years, the concept of CAR T-cells has been used in the development of other cell-based immunotherapies. According to the updated Cancer Cell Immunotherapy Pipeline 2022 from Cancer Research Institute, the number of active cell therapies developed in 2022 including CAR T-cells, TCR T-cells, NK and NKT cells, TIL cells, and tumor-associated antigen (TAA)/tumor-specific antigen (TSA) targeted T cells from preclinical to clinical trials and marketing is 2756, compared to 2031 in 2021. Of these, 1432 are active CAR T-cell therapies, 280 more than 1150 in 2021 [24]. Figure 2 illustrates the changes in the number of active CAR T-cell therapies in the past two years.
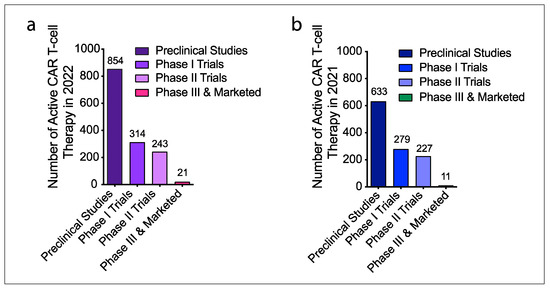
Figure 2. Active CAR T-cell therapies developed in 2022 and 2021. (a) A total of 1432 active CAR T-cell therapies have been developed in 2022, including preclinical research (854), phase I (314), phase II (243), phase III and market trials of 21. (b) A total of 1150 active CAR T-cell therapies were developed in 2021, including 633 preclinical research, 279 phase I, 227 phase II, 11 phase III and market trials.
However, CAR T-cell therapy has been disappointing in the treatment of solid tumors and faces many challenges. Currently, a single antigen-targeted CAR T-cell therapy for solid tumors frequently fails, and no dual antigens-targeted CAR T-cell therapies have been approved for marketing yet in the world. To overcome tumor-defense mechanisms including immunosuppression (immunosuppressive cytokines secreted by solid tumors), antigen escape, and physical barriers to infiltrate into solid tumors, more sophisticated engineering approaches are required to develop effective CAR T-cell therapies.
3. CAR T-Cell Therapy in Solid Tumors
With the promising results of CAR T-cell therapy in the treatment of hematological malignancies, scientists have begun to extend CAR T-cell therapy to metastatic solid tumors, including lung, ovarian, breast, prostate, liver, kidney, stomach, pancreatic, and colon cancer. However, response rates of CAR-T therapy in patients with solid tumors much lower than those with hematological malignancies. The ORR of CAR T-cell therapy in patients with solid malignancies was 20% (95% CI: 11–34%) vs. 71% (95% CI: 62–79%) in those with hematological malignancies. Disappointing CAR T-cell therapies in the treatment of solid tumors [25][26] indicate the potential issues in solid tumors as follows: (1) TAA antigen identification (particularly tumor-specific antigens), expression level, and susceptibility to CAR T-cells, (2) tumor infiltration, CAR T-cells may not be able to penetrate tumor tissue through the vascular endothelium, and (3) survival of CAR T-cells in TME.
In 2010, a patient with colon cancer metastasis to the lung and liver died after ERBB2-targeting CAR T-cell therapy [27]. The potential cause of death may be that CAR T-cells recognize low levels of ERBB2 on lung epithelial cells, thereby triggering cytokine storms.
In 2021, Tmunity Therapeutics, a clinical-stage biotherapeutics company, halted the development of its lead CAR T-cell product after the deaths of two patients in a clinical trial. The patients reportedly died from immune effector cell-associated neurotoxicity syndrome (ICANS) (Carroll J. Exclusive: Carl June’s Tmunity encounters a lethal roadblock as 2 patient deaths derail lead trial, raise red flag forcing rethink of CAR T-cells for solid tumors. Endpoints News. 2 June 2021. Accessed on 3 June 2021. https://bit.ly/3wPYWm0).
The greatest challenge of CAR T-cell therapy for solid tumors is to find a tumor antigen that is uniquely expressed on the surface of solid tumor cells to provide CAR T-cell specific target. However, a major limitation of CAR T-cell therapy is that most proteins are tumor-associated antigens (TAAs), which are also expressed at low levels in normal cells, making it difficult for CAR T-cells to specifically target tumor cells without impairing healthy cells. Additionally, CAR T-cells are limited in trafficking to and infiltrating solid tumors as an immunosuppressive tumor microenvironment and physical tumor barrier [28]. Current CAR T-cell products for solid tumors are single-target CAR T-cells, dual-target CAR T-cell therapies have not yet been approved for marketing.
CAR T-cell therapy shows a strong clinical efficacy in hematological malignancies with a complete remission (CR) of around 30–40% in treating advanced B-cell malignancies [29], but not yet in solid tumors except for some individual cases [30]. According to 2022 AACR, Haanen and colleagues conducted a clinical trial to evaluate the early efficacy and safety of the CAR T-cell product targeting CLDN6. Among the 14 patients who were evaluable at six weeks after infusion, 4 patients with testicular cancer and 2 with ovarian cancer experienced a partial response (PR), with an overall response rate of nearly 43% (https://www.aacr.org/about-the-aacr/newsroom/news-releases/new-car-t-cell-therapy-for-solid-tumors-was-safe-and-showed-early-efficacy/#:~:text=Among%20the%20study%20participants%20who,at%2012%20weeks%20after%20infusion (accessed on 18 November 2022)).
According to the updated data from Cancer Research Institute [24], the total number of active CAR T-cell therapies for solid tumors conducted in 2022 was 27. Of these 27 therapies, 4 CAR T-cell products target HER2, 6 target MSLN, 7 target GPC2/3, and 10 target EGFR. These newly developed T-cell therapies in solid tumor are summarized in Figure 3.
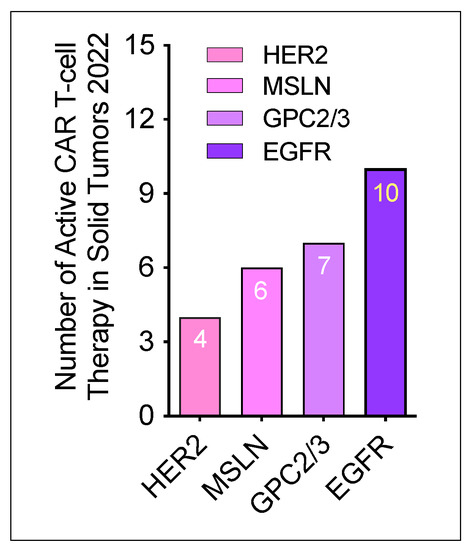
Figure 3. Number of active CAR T-cell trials investigated in 2022. Based on the different antigenic targets, 4 CAR T-cell products are anti-HER2, 6 CAR T-cell products are anti-4 MSLN, 7 CAR T-cell products are anti-4 GPC, 10 CAR T-cell products are anti-EGFR.
In addition, according to the information from the website of the Clinical Trials (https://www.clinicaltrials.gov (accessed on 25 October 2022)), more CAR T-cells are under investigation in phase I clinical trials for solid tumors. The efficacy of CAR T-cells in solid tumors has not been supported. The following are some main CAR T-cell products developed for the treatment of solid tumor. Except a few of them are discussed in the text based on their attention and clinical accidents in clinical trials, others cannot be described here due to the spatial limitation, but they are summarized in Table 1.
Table 1. Most targeted antigens in clinical trials of CAR T-cell therapy in solid tumors.
| Antigen | Cancer | Phase | Identifier (ID) |
| EGFR | Lung, colorectal, ovarian, pancreatic, renal cancers | Phase 1/2 | NCT01869166 |
| HER2 | Central nervous system tumor, pediatric glioma | Phase 1 | NCT03500991 |
| EGFR806 | Central nervous system tumor, pediatric glioma | Phase 1 | NCT03638167 |
| Mesothelin | Ovarian, cervical, pancreatic, lung | Phase 1/2 | NCT01583686 |
| PSCA | Lung | Phase 1 | NCT03198052 |
| MUC1 | Advanced solid tumors, lung | Phase 1/2 | NCT03179007, NCT03525782 |
| Claudin 18.2 | Advanced solid tumor | Phase 1 | NCT03874897 |
| EpCAM | Colon, pancreatic, prostate, gastric, liver | Phase 1/2 | NCT03013712 |
| GD2 | Brain | Phase 1 | NCT04099797 |
| VEGFR2 | Melanoma, brain | Phase 1 | NCT01218867 |
| AFP | Hepatocellular carcinoma liver cancer | Phase 1 | NCT03349255 |
| Nectin4/FAP | Nectin4-positive advanced malignant solid tumor | Phase 1 | NCT03932565 |
| CEA | Lung, colorectal, gastric, breast, pancreatic cancer | Phase 1 | NCT02349724 |
| Lewis Y | Advanced cancer | Phase 1 | NCT03851146 |
| Glypican-3 | Liver | Phase 1 | NCT02932956 |
| EGFRvIII | Glioblastoma and brain tumor | Phase 1 | NCT01454596 |
| IL-13Rα2 | Glioblastoma | Phase 1 | NCT02208362 |
| CD171 | Neuroblastoma | Phase 1 | NCT02311621 |
| MUC16 (CA-125) | Ovarian | Phase 1 | NCT 02498912 |
| PSMA | Prostate | Phase 1 | NCT01140373 |
| AFP | Hepatocellular carcinoma, liver | Phase 1 | NCT03349255 |
| AXL | Renal | Phase 1 | NCT03393936 |
| CD20 | Melanoma | Phase 1 | NCT03893019 |
| CD80/86 | Lung | Phase 1 | NCT03060343 |
| c-MET | Breast, hepatocellular | Phase 1 | NCT03060356, NCT03672305 |
| DLL-3 | Lung | Phase 1 | NCT03392064 |
| DR5 | Hepatoma | Phase 1 | NCT03638206 |
| EphA2 | Glioma | Phase 1 | NCT02575261 |
| TAG72 | Ovarian | Phase 1 | NCT05225363 |
| gp100 | Melanoma | Phase 1 | NCT03649529 |
| MAGE-A1/3/4 | Lung | Phase 1 | NCT03356808, NCT03535246 |
| LMP1 | Nasopharyngeal | Phase 1 | NCT02980315 |
Abbreviations: EGFR: epidermal growth factor receptor; HER2: human epidermal growth factor receptor 2; PSCA: prostate stem cell antigen; MUC1: mucin 1; EpCAM: epithelial cell adhesion molecule; GD2: disialoganglioside; VEGFR2: vascular endothelial growth factor receptor 2; AFP: alpha fetoprotein; FAP: fibroblast activation protein; CEA: carcinoembryonic antigen; IL-13R: interleukin-13 receptor; CD171: L1 cell adhesion molecule; MUC16: mucin 16; PSMA: prostate-specific membrane antigen; AXL: AXL receptor tyrosine kinase; c-MET: tyrosine-protein kinase Met; DLL-3: delta like canonical notch ligand 3; DR5: death receptor 5; EpHA2: ephrin type-A receptor 2; FR-α: Folate receptor alpha; gp100: glycoprotein 100; MAGE-A: melanoma-associated antigen 3; LMP1: latent membrane protein 1.
2.1. CAR T-Cells Targeting Prostate TAA
Prostate-specific membrane antigen (PSMA) is a type II integral membrane glycoprotein highly expressed in prostate cancer and is a diagnostic and prognostic marker, which is a tumor-associated antigen (TAA). PSMA levels in prostate cancer are 100 to 1000 times higher than in normal tissues [31]. Meanwhile, high levels of PSMA are associated with the aggressiveness of human malignancies [32][33]. In addition, PSMA is also highly expressed in tumor neovascularization [34]. Increased PSMA expression is an independent predictor of prostate cancer recurrence. Therefore, PSMA becomes an attractive new therapeutic target with the most minimal tissue penetration for the development of anti-PSMA CAR T-cell therapy in prostate cancer [35]. Anti-PSMA CAR T-cells have robust killing ability against human prostate cancer cells and demonstrated strong expansion and cytotoxicity potential in prostate cancer cells [36]. Clinical trials conducted by Junghans et al. [37] and Slovin et al. [38] confirmed the safety and efficacy of PSMA-targeted CAR T-cells for prostate cancer. However, PSMA is also expressed in benign prostatic epithelial cells and normal prostate tissue.
One CAR T-cell product targeting PSMA, P-PSMA-101, is from Poseida Therapeutics (San Diego, CA, USA), which was designed to target prostate cancer cells expressing the cell-surface antigen PSMA (https://poseida.com/science/pipeline (1 December 2022)). In preclinical studies, P-PSMA-101 has been shown to eliminate tumor cells to undetectable levels in 100% of animals, with only one incidence of relapse in the lower dose (NCT04249947). Based on published literature, no other product candidate has shown complete elimination of solid tumors in this preclinical model. A phase I clinical trial of P-PSMA-101 CAR T-cells in patients with metastatic castration resistant prostate cancer (mCRPC) has been conducted and a dose escalation trial of P-PSMA-101 is ongoing.
Another CAR T-cell product targeting PSMA, TmPSA01, is from Tmunity Therapeutics (Philadelphia, PA, USA), which is a pioneer and lead CAR T-cell product, which is at the forefront of advancing CAR T-cell applications. Unfortunately, TmPSA01 has been halted after two patients died of neurotoxicity. This work identified potential barriers to CAR T-cell therapy for solid tumors. Notably, the researchers identified cases of ICANS. Details of the news were first reported by endpoints and confirmed by Tmunity. This news has profound implications for the broader push to toward cell therapy as a treatment for solid tumors (https://www.fiercebiotech.com/biotech/tmunity-stops-solid-tumor-car-t-trial-after-2-patients-die (accessed on 30 October 2022)). At Tmunity, the setback has led to the end of the CART-PSMA-TGFβRDN study and the start of work on subsequent candidates with improved safety profile. Tmunity is aiming to file an IND in the second half of the year.
In addition to PSMA, prostate stem cell antigen (PSCA), another TAA for prostate cancer, is a membrane glycoprotein predominantly expressed in prostate cancer, which is expressed in 94% (105/112) of primary prostate tumors and 100% (9/9) of bone metastases [39]. In vivo studies have showed that anti-PSCA monoclonal antibodies inhibit tumor growth and metastasis formation, making PSCA potentially useful in immunotherapy programs for the treatment of prostate cancer [40][41]. Although the expression of PSCA is upregulated in most of prostate cancers, its biological role in prostate cancer remains unclear.
2.2. CAR T-Cells Targeting MSLN (ATA2271)
Mesothelin (MSLN) is a cell-surface antigen associated with tumor invasion, which is strongly expressed in many solid tumor types, including mesothelioma, lung cancer, breast cancer, and pancreatic cancer [42]. MSLN has emerged as an important target in CAR T-cell therapy. Phase I clinical trials have shown that MSLN-targeted CAR T-cell therapy is safe, but its efficacy is very limited due to insufficient tumor infiltration and the persistence of CAR T-cells [43]. Lv et al. constructed MSLN CAR T-cells using MSLN scFv, CD3ζ, CD28, and DAP10 intracellular signaling domain (M28z10) to target MSLN. The results of in vitro experiments showed that M28z10 T cells exhibited strong cytotoxicity and cytokine-secreting ability to gastric cancer cells. The in vivo experimental results showed that M28z10 T cells could induce gastric cancer regression and prolong the mouse survival in different xenograft mouse models [44].
The expression rate of MSLN was various among pathological types of solid cancer (serous 97%, clear cell 83%, endometrioid 77%, mucinous 71%, carcinosarcoma 65%), pancreatic adenocarcinoma (ductal 75%, ampullary 81%), endometrial carcinoma (clear cell 71%, serous 57%, carcinosarcoma 50%, endometrioid 45%), malignant mesothelioma (69%) and lung adenocarcinoma (55%) [45]. The highest prevalence of positive MSLN was found in ovarian cancer. Kachala et al. reported that MSLN overexpression is a tumor aggressive marker and is associated with increased risk of recurrence and decreased overall survival (OS) [46]. MSLN CAR T-cell therapy has the potential to treat a variety of solid malignancies that are overexpressed MSLN [42]. Schoutrop et al. evaluated the efficacy of MSLN-directed CAR T-cell therapy in an orthotopic mouse model of ovarian cancer. The results showed that MSLN CAR T-cell therapy significantly prolonged survival, but sustained tumor control was not observed [47].
Malignant pleural mesothelioma (MPM) is a rare but highly aggressive malignancy with limited treatment options [48]. It is characterized by resistance to treatment and poor survival [49]. The median OS of patients with MPM after the first-line treatment with cisplatin and pemetrexed was only 13 to 16 months, which was prolonged to 18.8 months after an addition of bevacizumab, but at the cost of increased toxicity [50]. MPM treatment guidelines from the National Comprehensive Cancer Network (NCCN) include the use of ICIs as the second-line treatment. Although the expression levels of PD-L1 and tumor mutation burden (TMB) are very low in patients with MPM [51], the responses still occurred to PD-L1 blockade [48]. The scientists at Memorial Sloan Kettering Cancer Center (MSKCC) developed and conducted the first-in-human phase I study of a regional, autologous, MSLN-targeted CAR T-cell therapy [52]. The results showed that intrapleural administration of 0.3 to 60 M mesothelin-targeted CAR T-cells/kg was safe and well tolerated in 27 patients (25 with MPM, one with metastatic lung cancer, another with metastatic breast cancer), and CAR T-cells were detected in peripheral blood for >100 days in 39% of patients. The median OS of patients receiving CAR T-cell infusion was 23.9 months (83% 1-year OS rate). Eight patients had a stable condition for ≥6 months; two patients showed complete metabolic responses after positron emission tomography (PET) scanning [52].
In addition to the traditional therapies described above, MSLN-targeted CAR T-cell therapy for patients with advanced mesothelioma using next-generation PD1DNR and 1XX CAR technology is also being tested in clinical trials. ATA2271, a next-generation autologous CAR T-cell therapy targeting MSLN, manufactured by Atara Biotherapeutics, Inc. (San Francisco, CA, USA) is currently under clinical investigation in patients with MPM. According to Atara Biotherapeutics, ATA2271 targets hard-to-treat solid tumors using proprietary 1XX CAR signaling and intrinsic PD-1 checkpoint inhibition.
An ongoing Phase 1 dose-escalation trial of advanced mesothelioma has demonstrated the early safety and durability of armored CAR T-cells in patients. The preliminary results of the next generation autologous MSLN-targeted CAR T-cell ATA2271 were presented at 2021ESMO Immuno-Oncology Conference (9 December 2021). But on 18 February 2022, scientists at MSKCC notified the FDA of a fatal serious adverse event (SAE) associated with a patient treated with autologous CAR T-cells.
According to the press release issued by Atala Biotherapy on 28 February 2022 (https://investors.atarabio.com/news-events/press-releases/detail/265/atara-biotherapeutics-provides-update-on-ata2271-autologous (18 November 2022)): the first 6 patients enrolled in the two lowest dose groups received either 1 × 106 cells/kg (patients 1–3) or 3 × 106 cells/kg (patient 4–6) of ATA2271 intrapleural treatment. No dose-limiting toxicities have been reported in either cohort. The reported patient event was related to the first patient in a third, higher-dose cohort (6 × 106 cells/kg). The temporary suspension of ATA2271 study enrollment does not affect the ongoing work to promote IND; ATA3271 is a separate, off-the-shelf, allogeneic ATA3271. ATA3219, Tabelecleucel (tab-cel), and ATA188 all utilize Atara’s allogeneic EBV T-cell platform, the safety and tolerability have been validated by clinical studies and experience in approximately 400 patients in various disease areas where CRS has not been observed to date.
The ongoing (from 30 September 2020 to September 2023) Phase 1 trial of MSLN-targeted CAR T-cell therapy in patients with mesothelioma is sponsored by MSKCC (https://clinicaltrials.gov/ct2/show/NCT04577326 (18 November 2022)). Intrapleural administration of ATA2271 was well-tolerated at the lowest dose levels, and no CAR T-cell related adverse events (AEs) of Grade > 2 observed and no AEs of Grade > 3 have been observed in the research to date. All four patients had received at least four prior lines of therapy. Importantly, ATA2271 CAR T-cells persisted in peripheral blood of patients for more than 4 weeks and were associated with upregulated effector cytokines.
2.3. Other Tumor Antigens Used for CAR T-Cells in Solid Tumors
2.3.1. MUC1
MUC1 is a transmembrane glycoprotein that is aberrantly glycosylated and overexpressed in a variety of epithelial cancers. Previous studies have confirmed that MUC1 is overexpressed in NSCLC tissues [53][54], and in about 70% of ovarian cancer [55]. Tumor-associated MUC1 (tMUC1) is different from the MUC1 expressed in normal cells and can be used as a biomarker and therapeutic target of cancer [56]. Zhou et al. reported that monoclonal antibody TAB004 specifically recognizes tMUC1 in all subtypes of breast cancer, including 95% of triple-negative breast cancer (TNBC), while retaining recognition of MUC1 in normal tissue [57]. The team transduced human T cells with MUC28z, a CAR comprised of the scFv of TAB004 coupled to CD28 and CD3ζ. The results showed that MUC28z was well expressed on the surface of engineered activated human T cells. MUC28z CAR T-cells showed significant target-specific cytotoxicity against a group of human TNBC cells.
2.3.2. ICAM1
Intercellular adhesion molecule-1 (ICAM1) is a cell surface transmembrane glycoprotein receptor. ICAM1 has been reported to be overexpressed in lung cancer, pancreatic cancer [58] and renal cell cancer [59]. High levels of ICAM1 were correlated with metastasis and poor prognosis in cancer patients [58]. ICAM1 expression is increased in TNBC patients and can be up to 200-fold increase in lung metastases of TNBC patients [60]. The authors demonstrated ICAM1 generated a phage-displayed scFv library using splenocytes from ICAM1-immunized mice and selected a novel ICAM1-specific scFv, mG2-scFv. Using mG2-scFv as the extracellular antigen binding domain, the team constructed ICAM1-specific CAR T-cells and demonstrated potent and specific killing of TNBC cell lines in vitro and in vivo [61].
2.3.3. EGFR
Epidermal growth factor receptor (EGFR) is a transmembrane protein involved in cell growth and differentiation. EGFR is overexpressed in a wide range of solid tumor types [62], it is critical to control the growth and survival of epithelial cells, including NSCLC [63]. EGFR targeted therapies includes tyrosine kinase inhibitors (TKIs, e.g., gefitinib and erlotinib, afatinib, Osimertinib) [64], phosphatidylinositol 3-kinase (PI3K) inhibitors, and antisense gene therapy. These EGFR TKIs have effectively replaced chemotherapy as the first line treatment [65], Unfortunately, EGFR is increasingly recognized as a biomarker of tumor resistance [62], since all patients with metastatic lung who initially benefit from EGFR-targeted therapies eventually developed resistance [66]. EGFR-specific CAR T-cells have been reported not only to trigger cell lysis of EGFR-positive TNBC in vitro, but also to inhibit the growth of mouse cell lines and patient-derived xenograft (PDX) TNBC tumors [67]. EGFR is also a target of immunotherapy [68][69]. EGFR monoclonal antibodies (mAbs, e.g., cetuximab, panitumumab, nimotuzumab, and necitumumab) have been developed for the treatment of cancer [70].
Li et al. showed that proliferation and anticancer effects of EGFR CAR T-cells in vitro depend on time (24 to 72 h) and antigen (with and without EGFR antigen stimulation), and the regression of EGFR-positive human lung cancer xenografts in vivo [71]. A phase I clinical trial of EGFR CAR T-cells (NCT03182816) demonstrated that EGFR CAR T- cell therapy was well tolerated in all nine patients in treatment of EGFR-positive advanced R/R NSCLC patients. The results showed that EGFR CAR T-cells were detectable in peripheral blood of eight patients, partial response (PR) was observed in one patient, stable disease (SD) in six patients, and progressive disease (PD) in two patients. The progression-free survival (PFS) of these 9 patients was 7.13 months (95% CI 2.71–17.10 months), and the median OS was 15.63 months (95% CI 8.82–22.03 months) [72].
2.3.4. ROR1
Receptor tyrosine kinase-like orphan receptor 1 (ROR1), a member of ROR family, is a protein encoded by ROR1 gene, and it is overexpressed in cancer [73]. For example, 28.6% was found in 56 histologically confirmed lung adenocarcinoma (using a cut-off of 1), or in 51.8% of the cases using the median value as threshold [73]. It was reported that ROR1 repression inhibits the growth of lung adenocarcinoma regardless of EGFR status, and leads to multiple acquired resistance mechanisms, including EGFR T790M, MET amplification and hepatocyte growth factor (HGF) overexpression [74]. ROR1 CAR T-cells can effectively kill lung cancer cells in a three-dimensional tumor model of NSCLC. Wallstabe et al. reported that ROR1 CART-cell treatment not only showed strong antitumor activity in human lung cancer cell line (A549), but also infiltrate into cancer tissue and eradicated multiple layers of tumor cells [75]. This result provides a new strategy for the clinical treatment of lung cancer. A clinical trial (NCT02706392) has been conducted to evaluate autologous ROR1 CAR T-cells in patients with advanced ROR1-positive and stage IV NSCLC; the results have not been released yet.
2.3.5. Trop2
Trophoblast cell surface antigen 2 (Trop2) is a widely expressed glycoprotein and a member of the epithelial cell adhesion molecule (EpCAM) family in many normal tissues, and overexpressed in a variety of human cancers, including gastric cancer [76] and breast cancer [77]. Trop2 has potential in promoting epithelial-mesenchymal transition (EMT) in human breast cancer [78]. Overexpression of Trop2 has prognostic significance [79]. A study demonstrated that intra-tumoral injection of bi-specific Trop2/PD-L1 CAR T-cells can significantly reduce the growth of gastric cancer, and the inhibitory effect is stronger than specific Trop2 CAR T-cells [80]. These results suggest that novel Trop2/PD-L1 CAR T-cells are involved in Trop2/PD-L1 and checkpoint blockade in gastric cancer, thereby promoting the cytotoxicity of CAR T-cells in gastric cancer and other types of solid tumors [80].
2.3.6. TAG72
Tumor-associated glycoprotein 72 (TAG-72) is a pan-adenocarcinoma oncofetal antigen that is highly expressed in ovarian cancers, and increased expression is associated with disease progression. The recurrence of ovarian cancer after surgery and multidrug chemotherapy is frequent, and novel therapeutic methods are urgently needed [81]. TAG72 has been used as a target for CAR T-cell therapy. Humanized TAG72-specific CAR T-cells have been reported to show potential cytotoxicity and cytokine production in ovarian cancer. On the other hand, TAG72-based CAR T cells significantly reduced the proliferative potential and improved the survival rate of mice [82]. Shu et al. demonstrated that the co-expression of the TAG-72 CAR and the CD47-truncated monomer CAR on T cells (dual CAR T-cell strategy) may be effective in ovarian cancer, and applicable to other adenocarcinomas [83].
2.3.7. CA9
Carbonic anhydrase IX (CA9/ or CA IX) is an enzyme encoded by the human CA9 gene [84]. CA IX is overexpressed in many types of cancer, including clear cell renal cell carcinoma (RCC) [85][86][87], cervical cancer [88], and breast and lung cancer, and CA IX promotes tumor growth by enhancing tumor acidosis [89] as other CA family members [90]. CAIX is a highly expressed on the surface of tumor cells in RCC [85]; thus, CAIX is a potential therapeutic target. CAIX overexpression increased the expression of 6-Phosphofructo-2-Kinase/Fructose-2, 6-Biphosphatase 4 (PFKFB4) and EMT, and promoted the migration of cervical cancer cells. CAIX can promote metastasis of cervical cancer cells, thus its inhibitory effect can be used as a therapeutic strategy for cervical cancer [88]. Li et al. reported that CAIX CAR T-cells combined with sunitinib induced an effective antitumor response in an experimental model of metastatic RCC [91].
2.3.8. CD133
CD133 (Prominin 1, PROM1) is a transmembrane protein whose mRNA and glycosylated forms are highly expressed in a variety of human cancer cells. CD133 is a cancer stem cell (CSC) marker and is associated with cancer progression and patient prognosis [92][93], including pancreatic cancer [94], colorectal cancer [95] and breast cancer [96][97]. Besides the application of CD133-targeted CAR T-cells in MLL leukemia [98], anti-CD133 CAR T-cells have been reported in a phase I trial including 14 patients with hepatocellular carcinoma (HCC), 7 patients with pancreatic carcinomas, and 2 patients with colorectal cancer [99]. The results demonstrated the feasibility, controllable toxicities, and efficacy of anti-CD133 CAR T-cell therapy, with 3 patients achieving PR and 14 patients achieving SD among the 23 patients enrolled.
2.3.9. Integrin αvβ6
Integrin αvβ6 is an exciting biomarker and therapeutic target for pancreatic cancer, and it is highly expressed in almost 100% of pancreatic ductal adenocarcinoma (PDAC) cases [100]. It was reported that CAR T-cells expressing CXCR2 exhibited stronger anti-tumor activity against pancreatic tumor xenografts known to express αvβ6 [101].
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33.
- Yao, L.; Jia, G.; Lu, L.; Bao, Y.; Ma, W. Factors affecting tumor responders and predictive biomarkers of toxicities in cancer patients treated with immune checkpoint inhibitors. Int. Immunopharmacol. 2020, 85, 106628.
- He, X.; Xu, C. Immune checkpoint signaling and cancer immunotherapy. Cell. Res. 2020, 30, 660–669.
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801.
- Flego, M.; Frau, A.; Accardi, L.; Mallano, A.; Ascione, A.; Gellini, M.; Fanunza, E.; Vella, S.; Di Bonito, P.; Tramontano, E. Intracellular human antibody fragments recognizing the VP35 protein of Zaire Ebola filovirus inhibit the protein activity. BMC Biotechnol. 2019, 19, 64.
- Marofi, F.; Motavalli, R.; Safonov, V.A.; Thangavelu, L.; Yumashev, A.V.; Alexander, M.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; Jarahian, M.; et al. CAR T cells in solid tumors: Challenges and opportunities. Stem. Cell. Res. 2021, 12, 81.
- Ingegnere, T.; Mariotti, F.R.; Pelosi, A.; Quintarelli, C.; De Angelis, B.; Tumino, N.; Besi, F.; Cantoni, C.; Locatelli, F.; Vacca, P.; et al. Human CAR NK Cells: A New Non-viral Method Allowing High Efficient Transfection and Strong Tumor Cell Killing. Front. Immunol. 2019, 10, 957.
- Peinelt, A.; Bremm, M.; Kreyenberg, H.; Cappel, C.; Banisharif-Dehkordi, J.; Erben, S.; Rettinger, E.; Jarisch, A.; Meisel, R.; Schlegel, P.G.; et al. Monitoring of Circulating CAR T Cells: Validation of a Flow Cytometric Assay, Cellular Kinetics, and Phenotype Analysis Following Tisagenlecleucel. Front. Immunol. 2022, 13, 830773.
- Neelapu, S.S. CAR-T efficacy: Is conditioning the key? Blood 2019, 133, 1799–1800.
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, O.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554.
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544.
- Nastoupil, L.J.; Jain, M.D.; Feng, L.; Spiegel, J.Y.; Ghobadi, A.; Lin, Y.; Dahiya, S.; Lunning, M.; Lekakis, L.; Reagan, P.; et al. Standard-of-Care Axicabtagene Ciloleucel for Relapsed or Refractory Large B-Cell Lymphoma: Results From the US Lymphoma CAR T Consortium. J. Clin. Oncol. 2020, 38, 3119–3128.
- Gupta, A.; Gill, S. CAR-T cell persistence in the treatment of leukemia and lymphoma. Leuk. Lymphoma. 2021, 62, 2587–2599.
- Sarikonda, G.; Mathieu, M.; Natalia, M.; Pahuja, A.; Xue, Q.; Pierog, P.L.; Trampont, P.C.; Decman, V.; Reynolds, S.; Hanafi, L.A.; et al. Best practices for the development, analytical validation and clinical implementation of flow cytometric methods for chimeric antigen receptor T cell analyses. Cytom. B Clin. Cytom. 2021, 100, 79–91.
- Bar-Ephraim, Y.E.; Koning, J.J.; Burniol Ruiz, E.; Konijn, T.; Mourits, V.P.; Lakeman, K.A.; Boon, L.; Bogels, M.; van Maanen, J.P.; Den Haan, J.M.M.; et al. CD62L Is a Functional and Phenotypic Marker for Circulating Innate Lymphoid Cell Precursors. J. Immunol. 2019, 202, 171–182.
- Xu, C.; Yin, Y. Measuring Chimeric Antigen Receptor T Cells (CAR T Cells) Activation by Coupling Intracellular Cytokine Staining with Flow Cytometry. Methods. Mol. Biol. 2020, 2108, 159–165.
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733.
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 2013, 5, 177ra138.
- Menetrier-Caux, C.; Ray-Coquard, I.; Blay, J.Y.; Caux, C. Lymphopenia in Cancer Patients and its Effects on Response to Immunotherapy: An opportunity for combination with Cytokines? J. Immunother. Cancer 2019, 7, 85.
- Gutkin, P.M.; Kozak, M.M.; von Eyben, R.; Horst, K.C. Lymphopenia and clinical outcomes in patients with residual nodal disease after neoadjuvant chemotherapy for breast cancer. Cancer Causes Control 2020, 31, 1021–1026.
- Grossman, S.A.; Ellsworth, S.; Campian, J.; Wild, A.T.; Herman, J.M.; Laheru, D.; Brock, M.; Balmanoukian, A.; Ye, X. Survival in Patients With Severe Lymphopenia Following Treatment With Radiation and Chemotherapy for Newly Diagnosed Solid Tumors. J. Natl. Compr. Canc. Netw. 2015, 13, 1225–1231.
- Fabrizio, V.A.; Boelens, J.J.; Mauguen, A.; Baggott, C.; Prabhu, S.; Egeler, E.; Mavroukakis, S.; Pacenta, H.; Phillips, C.L.; Rossoff, J.; et al. Optimal fludarabine lymphodepletion is associated with improved outcomes after CAR T-cell therapy. Blood Adv. 2022, 6, 1961–1968.
- Pietrobon, V.; Todd, L.A.; Goswami, A.; Stefanson, O.; Yang, Z.; Marincola, F. Improving CAR T-Cell Persistence. Int. J. Mol. Sci. 2021, 22, 10828.
- Saez-Ibanez, A.R.; Upadhaya, S.; Partridge, T.; Shah, M.; Correa, D.; Campbell, J. Landscape of cancer cell therapies: Trends and real-world data. Nat. Rev. Drug Discov. 2022, 21, 631–632.
- Newick, K.; Moon, E.; Albelda, S.M. Chimeric antigen receptor T-cell therapy for solid tumors. Mol. Oncolytics. 2016, 3, 16006.
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73.
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851.
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69.
- Jenkins, Y.; Zabkiewicz, J.; Ottmann, O.; Jones, N. Tinkering under the Hood: Metabolic Optimisation of CAR-T Cell Therapy. Antibodies 2021, 10, 17.
- Weber, E.W.; Maus, M.V.; Mackall, C.L. The Emerging Landscape of Immune Cell Therapies. Cell 2020, 181, 46–62.
- Pianou, N.K.; Stavrou, P.Z.; Vlontzou, E.; Rondogianni, P.; Exarhos, D.N.; Datseris, I.E. More advantages in detecting bone and soft tissue metastases from prostate cancer using (18)F-PSMA PET/CT. Hell. J. Nucl. Med. 2019, 22, 6–9.
- Kessel, K.; Bernemann, C.; Bogemann, M.; Rahbar, K. Evolving Castration Resistance and Prostate Specific Membrane Antigen Expression: Implications for Patient Management. Cancers 2021, 13, 3556.
- Chang, S.S. Overview of prostate-specific membrane antigen. Rev. Urol. 2004, 6 (Suppl. 10), S13–S18.
- Van de Wiele, C.; Sathekge, M.; de Spiegeleer, B.; De Jonghe, P.J.; Debruyne, P.R.; Borms, M.; Beels, L.; Maes, A. PSMA expression on neovasculature of solid tumors. Histol. Histopathol. 2020, 35, 919–927.
- Hillerdal, V.; Essand, M. Chimeric antigen receptor-engineered T cells for the treatment of metastatic prostate cancer. BioDrugs 2015, 29, 75–89.
- Weimin, S.; Abula, A.; Qianghong, D.; Wenguang, W. Chimeric cytokine receptor enhancing PSMA-CAR-T cell-mediated prostate cancer regression. Cancer Biol. Ther. 2020, 21, 570–580.
- Junghans, R.P.; Ma, Q.; Rathore, R.; Gomes, E.M.; Bais, A.J.; Lo, A.S.; Abedi, M.; Davies, R.A.; Cabral, H.J.; Al-Homsi, A.S.; et al. Phase I Trial of Anti-PSMA Designer CAR-T Cells in Prostate Cancer: Possible Role for Interacting Interleukin 2-T Cell Pharmacodynamics as a Determinant of Clinical Response. Prostate 2016, 76, 1257–1270.
- Slovin, S.F.; Wang, X.; Hullings, M.; Arauz, G.; Bartido, S.; Lewis, J.S.; Schöder, H.; Zanzonico, P.; Scher, H.I.; Riviere, I. Chimeric antigen receptor (CAR+) modified T cells targeting prostate specific membrane antigen (PSMA) in patients (pts) with castrate metastatic prostate cancer (CMPC). J. Clin. Oncol. 2013, 31, 72.
- Gu, Z.; Thomas, G.; Yamashiro, J.; Shintaku, I.P.; Dorey, F.; Raitano, A.; Witte, O.N.; Said, J.W.; Loda, M.; Reiter, R.E. Prostate stem cell antigen (PSCA) expression increases with high gleason score, advanced stage and bone metastasis in prostate cancer. Oncogene 2000, 19, 1288–1296.
- Yu, S.; Feng, F.; Wang, K.; Men, C.; Lin, C.; Liu, Q.; Yang, D.; Gao, Z. The therapeutic efficacy of I131-PSCA-mAb in orthotopic mouse models of prostate cancer. Eur. J. Med. Res. 2013, 18, 56.
- Dannull, J.; Diener, P.A.; Prikler, L.; Furstenberger, G.; Cerny, T.; Schmid, U.; Ackermann, D.K.; Groettrup, M. Prostate stem cell antigen is a promising candidate for immunotherapy of advanced prostate cancer. Cancer Res. 2000, 60, 5522–5528.
- Morello, A.; Sadelain, M.; Adusumilli, P.S. Mesothelin-Targeted CARs: Driving T Cells to Solid Tumors. Cancer Discov. 2016, 6, 133–146.
- Klampatsa, A.; Dimou, V.; Albelda, S.M. Mesothelin-targeted CAR-T cell therapy for solid tumors. Expert. Opin. Biol. Ther. 2021, 21, 473–486.
- Lv, J.; Zhao, R.; Wu, D.; Zheng, D.; Wu, Z.; Shi, J.; Wei, X.; Wu, Q.; Long, Y.; Lin, S.; et al. Mesothelin is a target of chimeric antigen receptor T cells for treating gastric cancer. J. Hematol. Oncol. 2019, 12, 18.
- Weidemann, S.; Gagelmann, P.; Gorbokon, N.; Lennartz, M.; Menz, A.; Luebke, A.M.; Kluth, M.; Hube-Magg, C.; Blessin, N.C.; Fraune, C.; et al. Mesothelin Expression in Human Tumors: A Tissue Microarray Study on 12,679 Tumors. Biomedicines 2021, 9, 397.
- Kachala, S.S.; Bograd, A.J.; Villena-Vargas, J.; Suzuki, K.; Servais, E.L.; Kadota, K.; Chou, J.; Sima, C.S.; Vertes, E.; Rusch, V.W.; et al. Mesothelin overexpression is a marker of tumor aggressiveness and is associated with reduced recurrence-free and overall survival in early-stage lung adenocarcinoma. Clin. Cancer Res. 2014, 20, 1020–1028.
- Schoutrop, E.; El-Serafi, I.; Poiret, T.; Zhao, Y.; Gultekin, O.; He, R.; Moyano-Galceran, L.; Carlson, J.W.; Lehti, K.; Hassan, M.; et al. Mesothelin-Specific CAR T Cells Target Ovarian Cancer. Cancer Res. 2021, 81, 3022–3035.
- Raghav, K.; Liu, S.; Overman, M.J.; Willett, A.F.; Knafl, M.; Fu, S.C.; Malpica, A.; Prasad, S.; Royal, R.E.; Scally, C.P.; et al. Efficacy, Safety, and Biomarker Analysis of Combined PD-L1 (Atezolizumab) and VEGF (Bevacizumab) Blockade in Advanced Malignant Peritoneal Mesothelioma. Cancer Discov. 2021, 11, 2738–2747.
- Vogelzang, N.J.; Rusthoven, J.J.; Symanowski, J.; Denham, C.; Kaukel, E.; Ruffie, P.; Gatzemeier, U.; Boyer, M.; Emri, S.; Manegold, C.; et al. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J. Clin. Oncol. 2003, 21, 2636–2644.
- Zalcman, G.; Mazieres, J.; Margery, J.; Greillier, L.; Audigier-Valette, C.; Moro-Sibilot, D.; Molinier, O.; Corre, R.; Monnet, I.; Gounant, V.; et al. Bevacizumab for newly diagnosed pleural mesothelioma in the Mesothelioma Avastin Cisplatin Pemetrexed Study (MAPS): A randomised, controlled, open-label, phase 3 trial. Lancet 2016, 387, 1405–1414.
- Yarchoan, M.; Albacker, L.A.; Hopkins, A.C.; Montesion, M.; Murugesan, K.; Vithayathil, T.T.; Zaidi, N.; Azad, N.S.; Laheru, D.A.; Frampton, G.M.; et al. PD-L1 expression and tumor mutational burden are independent biomarkers in most cancers. JCI Insight 2019, 4, e126908.
- Adusumilli, P.S.; Zauderer, M.G.; Riviere, I.; Solomon, S.B.; Rusch, V.W.; O’Cearbhaill, R.E.; Zhu, A.; Cheema, W.; Chintala, N.K.; Halton, E.; et al. A Phase I Trial of Regional Mesothelin-Targeted CAR T-cell Therapy in Patients with Malignant Pleural Disease, in Combination with the Anti-PD-1 Agent Pembrolizumab. Cancer Discov. 2021, 11, 2748–2763.
- Saltos, A.; Khalil, F.; Smith, M.; Li, J.; Schell, M.; Antonia, S.J.; Gray, J.E. Clinical associations of mucin 1 in human lung cancer and precancerous lesions. Oncotarget 2018, 9, 35666–35675.
- Tu, J.; Tang, M.; Li, G.; Chen, L.; Wang, Y.; Huang, Y. Expression of Mucin Family Proteins in Non-Small-Cell Lung Cancer and its Role in Evaluation of Prognosis. J. Oncol. 2022, 2022, 4181658.
- Patel, U.; Abernathy, J.; Savani, B.N.; Oluwole, O.; Sengsayadeth, S.; Dholaria, B. CAR T cell therapy in solid tumors: A review of current clinical trials. EJHaem 2022, 3, 24–31.
- Nath, S.; Mukherjee, P. MUC1: A multifaceted oncoprotein with a key role in cancer progression. Trends. Mol. Med. 2014, 20, 332–342.
- Zhou, R.; Yazdanifar, M.; Roy, L.D.; Whilding, L.M.; Gavrill, A.; Maher, J.; Mukherjee, P. CAR T Cells Targeting the Tumor MUC1 Glycoprotein Reduce Triple-Negative Breast Cancer Growth. Front. Immunol. 2019, 10, 1149.
- Huang, C.; Li, N.; Li, Z.; Chang, A.; Chen, Y.; Zhao, T.; Li, Y.; Wang, X.; Zhang, W.; Wang, Z.; et al. Tumour-derived Interleukin 35 promotes pancreatic ductal adenocarcinoma cell extravasation and metastasis by inducing ICAM1 expression. Nat. Commun. 2017, 8, 14035.
- Tanabe, K.; Campbell, S.C.; Alexander, J.P.; Steinbach, F.; Edinger, M.G.; Tubbs, R.R.; Novick, A.C.; Klein, E.A. Molecular regulation of intercellular adhesion molecule 1 (ICAM-1) expression in renal cell carcinoma. Urol. Res. 1997, 25, 231–238.
- Taftaf, R.; Liu, X.; Singh, S.; Jia, Y.; Dashzeveg, N.K.; Hoffmann, A.D.; El-Shennawy, L.; Ramos, E.K.; Adorno-Cruz, V.; Schuster, E.J.; et al. ICAM1 initiates CTC cluster formation and trans-endothelial migration in lung metastasis of breast cancer. Nat. Commun. 2021, 12, 4867.
- Wei, H.; Wang, Z.; Kuang, Y.; Wu, Z.; Zhao, S.; Zhang, Z.; Li, H.; Zheng, M.; Zhang, N.; Long, C.; et al. Intercellular Adhesion Molecule-1 as Target for CAR-T-Cell Therapy of Triple-Negative Breast. Cancer Front. Immunol. 2020, 11, 573823.
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20.
- Yu, J.J.; Zhou, D.D.; Yang, X.X.; Cui, B.; Tan, F.W.; Wang, J.; Li, K.; Shang, S.; Zhang, C.; Lv, X.X.; et al. TRIB3-EGFR interaction promotes lung cancer progression and defines a therapeutic target. Nat. Commun. 2020, 11, 3660.
- Tan, C.S.; Kumarakulasinghe, N.B.; Huang, Y.Q.; Ang, Y.L.E.; Choo, J.R.; Goh, B.C.; Soo, R.A. Third generation EGFR TKIs: Current data and future directions. Mol. Cancer 2018, 17, 29.
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454.
- Chong, C.R.; Janne, P.A. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat. Med. 2013, 19, 1389–1400.
- Liu, Y.; Zhou, Y.; Huang, K.H.; Li, Y.; Fang, X.; An, L.; Wang, F.; Chen, Q.; Zhang, Y.; Shi, A.; et al. EGFR-specific CAR-T cells trigger cell lysis in EGFR-positive TNBC. Aging 2019, 11, 11054–11072.
- Santos, E.D.S.; Nogueira, K.A.B.; Fernandes, L.C.C.; Martins, J.R.P.; Reis, A.V.F.; Neto, J.B.V.; Junior, I.; Pessoa, C.; Petrilli, R.; Eloy, J.O. EGFR targeting for cancer therapy: Pharmacology and immunoconjugates with drugs and nanoparticles. Int. J. Pharm. 2021, 592, 120082.
- Diaz-Serrano, A.; Gella, P.; Jimenez, E.; Zugazagoitia, J.; Paz-Ares Rodriguez, L. Targeting EGFR in Lung Cancer: Current Standards and Developments. Drugs 2018, 78, 893–911.
- Martinelli, E.; De Palma, R.; Orditura, M.; De Vita, F.; Ciardiello, F. Anti-epidermal growth factor receptor monoclonal antibodies in cancer therapy. Clin. Exp. Immunol. 2009, 158, 1–9.
- Li, H.; Huang, Y.; Jiang, D.Q.; Cui, L.Z.; He, Z.; Wang, C.; Zhang, Z.W.; Zhu, H.L.; Ding, Y.M.; Li, L.F.; et al. Antitumor activity of EGFR-specific CAR T cells against non-small-cell lung cancer cells in vitro and in mice. Cell. Death. Dis. 2018, 9, 177.
- Zhang, Y.; Zhang, Z.; Ding, Y.; Fang, Y.; Wang, P.; Chu, W.; Jin, Z.; Yang, X.; Wang, J.; Lou, J.; et al. Phase I clinical trial of EGFR-specific CAR-T cells generated by the piggyBac transposon system in advanced relapsed/refractory non-small cell lung cancer patients. J. Cancer Res. Clin. Oncol. 2021, 147, 3725–3734.
- Schiavone, G.; Epistolio, S.; Martin, V.; Molinari, F.; Barizzi, J.; Mazzucchelli, L.; Frattini, M.; Wannesson, L. Functional and clinical significance of ROR1 in lung adenocarcinoma. BMC Cancer 2020, 20, 1085, Erratum in BMC Cancer 2020, 20, 1196.
- Karachaliou, N.; Gimenez-Capitan, A.; Drozdowskyj, A.; Viteri, S.; Moran, T.; Carcereny, E.; Massuti, B.; Vergnenegre, A.; de Marinis, F.; Molina, M.A.; et al. ROR1 as a novel therapeutic target for EGFR-mutant non-small-cell lung cancer patients with the EGFR T790M mutation. Transl. Lung. Cancer Res. 2014, 3, 122–130.
- Wallstabe, L.; Gottlich, C.; Nelke, L.C.; Kuhnemundt, J.; Schwarz, T.; Nerreter, T.; Einsele, H.; Walles, H.; Dandekar, G.; Nietzer, S.L.; et al. ROR1-CAR T cells are effective against lung and breast cancer in advanced microphysiologic 3D tumor models. JCI Insight 2019, 4, e126345.
- Zhao, W.; Zhu, H.; Zhang, S.; Yong, H.; Wang, W.; Zhou, Y.; Wang, B.; Wen, J.; Qiu, Z.; Ding, G.; et al. Trop2 is overexpressed in gastric cancer and predicts poor prognosis. Oncotarget 2016, 7, 6136–6145.
- Zhu, J.; Wu, W.; Togashi, Y.; Taira Nihira, N.; Johmura, Y.; Zhu, D.; Nakanishi, M.; Miyoshi, Y.; Ohta, T. Alteration of Trop-2 expression in breast cancer cells by clinically used therapeutic agents and acquired tamoxifen resistance. Breast Cancer 2022, 29, 1076–1087.
- Zhao, W.; Kuai, X.; Zhou, X.; Jia, L.; Wang, J.; Yang, X.; Tian, Z.; Wang, X.; Lv, Q.; Wang, B.; et al. Trop2 is a potential biomarker for the promotion of EMT in human breast cancer. Oncol. Rep. 2018, 40, 759–766.
- Shvartsur, A.; Bonavida, B. Trop2 and its overexpression in cancers: Regulation and clinical/therapeutic implications. Genes Cancer 2015, 6, 84–105.
- Zhao, W.; Jia, L.; Zhang, M.; Huang, X.; Qian, P.; Tang, Q.; Zhu, J.; Feng, Z. The killing effect of novel bi-specific Trop2/PD-L1 CAR-T cell targeted gastric cancer. Am. J. Cancer Res. 2019, 9, 1846–1856.
- Ponnusamy, M.P.; Venkatraman, G.; Singh, A.P.; Chauhan, S.C.; Johansson, S.L.; Jain, M.; Smith, L.; Davis, J.S.; Remmenga, S.W.; Batra, S.K. Expression of TAG-72 in ovarian cancer and its correlation with tumor stage and patient prognosis. Cancer Lett. 2007, 251, 247–257.
- Murad, J.P.; Kozlowska, A.K.; Lee, H.J.; Ramamurthy, M.; Chang, W.C.; Yazaki, P.; Colcher, D.; Shively, J.; Cristea, M.; Forman, S.J.; et al. Effective Targeting of TAG72(+) Peritoneal Ovarian Tumors via Regional Delivery of CAR-Engineered T Cells. Front. Immunol. 2018, 9, 2268.
- Shu, R.; Evtimov, V.J.; Hammett, M.V.; Nguyen, N.N.; Zhuang, J.; Hudson, P.J.; Howard, M.C.; Pupovac, A.; Trounson, A.O.; Boyd, R.L. Engineered CAR-T cells targeting TAG-72 and CD47 in ovarian cancer. Mol. Ther. Oncolytics 2021, 20, 325–341.
- Nakagawa, Y.; Uemura, H.; Hirao, Y.; Yoshida, K.; Saga, S.; Yoshikawa, K. Radiation hybrid mapping of the human MN/CA9 locus to chromosome band 9p12-p13. Genomics 1998, 53, 118–119.
- Courcier, J.; de la Taille, A.; Nourieh, M.; Leguerney, I.; Lassau, N.; Ingels, A. Carbonic Anhydrase IX in Renal Cell Carcinoma, Implications for Disease Management. Int. J. Mol. Sci. 2020, 21, 7146.
- Genega, E.M.; Ghebremichael, M.; Najarian, R.; Fu, Y.; Wang, Y.; Argani, P.; Grisanzio, C.; Signoretti, S. Carbonic anhydrase IX expression in renal neoplasms: Correlation with tumor type and grade. Am. J. Clin. Pathol. 2010, 134, 873–879.
- Zhao, Z.; Liao, G.; Li, Y.; Zhou, S.; Zou, H.; Fernando, S. Prognostic value of carbonic anhydrase IX immunohistochemical expression in renal cell carcinoma: A meta-analysis of the literature. PLoS ONE 2014, 9, e114096.
- Hsin, M.C.; Hsieh, Y.H.; Hsiao, Y.H.; Chen, P.N.; Wang, P.H.; Yang, S.F. Carbonic Anhydrase IX Promotes Human Cervical Cancer Cell Motility by Regulating PFKFB4 Expression. Cancers 2021, 13, 1174.
- Chiche, J.; Brahimi-Horn, M.C.; Pouyssegur, J. Tumour hypoxia induces a metabolic shift causing acidosis: A common feature in cancer. J. Cell. Mol. Med. 2010, 14, 771–794.
- Han, R.; Yang, H.; Lu, L.; Lin, L. Tiliroside as a CAXII inhibitor suppresses liver cancer development and modulates E2Fs/Caspase-3 axis. Sci. Rep. 2021, 11, 8626.
- Li, H.; Ding, J.; Lu, M.; Liu, H.; Miao, Y.; Li, L.; Wang, G.; Zheng, J.; Pei, D.; Zhang, Q. CAIX-specific CAR-T Cells and Sunitinib Show Synergistic Effects Against Metastatic Renal Cancer Models. J. Immunother. 2020, 43, 16–28.
- Wang, H.; Gong, P.; Li, J.; Fu, Y.; Zhou, Z.; Liu, L. Role of CD133 in human embryonic stem cell proliferation and teratoma formation. Stem. Cell. Res. Ther. 2020, 11, 208.
- Ehteram, H.; Aslanbeigi, F.; Ghoochani Khorasani, E.; Tolouee, M.; Haddad Kashani, H. Expression and Prognostic Significance of Stem Cell Marker CD133 in Survival Rate of Patients with Colon Cancer. Oncol. Ther. 2022, 10, 451–461.
- Nomura, A.; Banerjee, S.; Chugh, R.; Dudeja, V.; Yamamoto, M.; Vickers, S.M.; Saluja, A.K. CD133 initiates tumors, induces epithelial-mesenchymal transition and increases metastasis in pancreatic cancer. Oncotarget 2015, 6, 8313–8322.
- Tsunekuni, K.; Konno, M.; Haraguchi, N.; Koseki, J.; Asai, A.; Matsuoka, K.; Kobunai, T.; Takechi, T.; Doki, Y.; Mori, M.; et al. CD44/CD133-Positive Colorectal Cancer Stem Cells are Sensitive to Trifluridine Exposure. Sci. Rep. 2019, 9, 14861.
- Brugnoli, F.; Grassilli, S.; Al-Qassab, Y.; Capitani, S.; Bertagnolo, V. CD133 in Breast Cancer Cells: More than a Stem Cell Marker. J. Oncol. 2019, 2019, 7512632.
- Han, R.; Yang, H.; Ling, C.; Lu, L. Neurospora crassa is a potential source of anti-cancer agents against breast cancer. Breast Cancer 2022, 29, 1032–1041.
- Bueno, C.; Velasco-Hernandez, T.; Gutierrez-Aguera, F.; Zanetti, S.R.; Baroni, M.L.; Sanchez-Martinez, D.; Molina, O.; Closa, A.; Agraz-Doblas, A.; Marin, P.; et al. CD133-directed CAR T-cells for MLL leukemia: On-target, off-tumor myeloablative toxicity. Leukemia 2019, 33, 2090–2125.
- Wang, Y.; Chen, M.; Wu, Z.; Tong, C.; Dai, H.; Guo, Y.; Liu, Y.; Huang, J.; Lv, H.; Luo, C.; et al. CD133-directed CAR T cells for advanced metastasis malignancies: A phase I trial. Oncoimmunology 2018, 7, e1440169.
- Reader, C.S.; Vallath, S.; Steele, C.W.; Haider, S.; Brentnall, A.; Desai, A.; Moore, K.M.; Jamieson, N.B.; Chang, D.; Bailey, P.; et al. The integrin alphavbeta6 drives pancreatic cancer through diverse mechanisms and represents an effective target for therapy. J. Pathol. 2019, 249, 332–342.
- Whilding, L.M.; Halim, L.; Draper, B.; Parente-Pereira, A.C.; Zabinski, T.; Davies, D.M.; Maher, J. CAR T-Cells Targeting the Integrin alphavbeta6 and Co-Expressing the Chemokine Receptor CXCR2 Demonstrate Enhanced Homing and Efficacy against Several Solid Malignancies. Cancers 2019, 11, 674.
More
Information
Subjects:
Immunology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
2 times
(View History)
Update Date:
16 Dec 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No