Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Martha Legorreta-Herrera | -- | 2232 | 2022-12-14 23:18:06 | | | |
| 2 | Vivi Li | Meta information modification | 2232 | 2022-12-15 02:18:31 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Buendía-González, F.O.; Legorreta-Herrera, M. Testosterone and DHEA and Immune Response. Encyclopedia. Available online: https://encyclopedia.pub/entry/38788 (accessed on 01 August 2026).
Buendía-González FO, Legorreta-Herrera M. Testosterone and DHEA and Immune Response. Encyclopedia. Available at: https://encyclopedia.pub/entry/38788. Accessed August 01, 2026.
Buendía-González, Fidel Orlando, Martha Legorreta-Herrera. "Testosterone and DHEA and Immune Response" Encyclopedia, https://encyclopedia.pub/entry/38788 (accessed August 01, 2026).
Buendía-González, F.O., & Legorreta-Herrera, M. (2022, December 14). Testosterone and DHEA and Immune Response. In Encyclopedia. https://encyclopedia.pub/entry/38788
Buendía-González, Fidel Orlando and Martha Legorreta-Herrera. "Testosterone and DHEA and Immune Response." Encyclopedia. Web. 14 December, 2022.
Copy Citation
Androgens are steroids that modulate various processes in the body, ranging from reproduction, metabolism, and even immune response. The main androgens are testosterone, dihydrotestosterone (DHT) and dehydroepiandrosterone (DHEA). These steroids modulate the development and function of immune response cells. Androgens are generally attributed to immunosuppressive effects; however, this is not always the case. Variations in the concentrations of these hormones induce differences in the innate, humoral, and cell-mediated immune response, which is concentration dependent. The androgens at the highest concentration in the organism that bind to the androgen receptor (AR) are DHEA and testosterone.
testosterone
DHEA
DHT
orchidectomy
gonadectomy
dimorphism
male
androgen receptor (AR)
1. Introduction
Androgens are steroid hormones with immunoregulatory properties, which interact with many cells of the immune system, both innate and adaptive immunity. These hormones modulate different responses in lymphoid and nonlymphoid tissues after interacting with the androgen receptor (AR). In general, androgens possess immunosuppressive properties, which at least partly explains the increased susceptibility of males compared to females to a variety of parasitic, bacterial, and viral infections [1][2][3][4][5].
There is evidence that androgens modulate the immune system. Testosterone and dehydroepiandrosterone (DHEA) are the androgens with the highest concentrations. The concentration of androgens is age and sex dependent, which complicates understanding their role in the immune response [6][7][8]. Understanding this phenomenon will contribute to explaining the greater susceptibility of males to bacterial, parasitic, and viral infections.
2. Synthesis of Androgens Testosterone, DHT, Androstenedione and DHEA and Its Main Properties
Synthesis of androgens starts with cholesterol [9][10], which is converted to pregnenolone through the action of the enzyme P450scc. This steroid in turn is converted to 17-OH pregnenolone by the enzyme 17α-hydroxylase. 17-OH pregnenolone is transformed to DHEA by the enzyme 17,20 lyase, and DHEA is in turn sulfated (DHEA-S) by the enzyme sulfotransferase, which keeps it stable for longer. DHEA and DHEA-S are the androgens with the highest concentrations in human blood circulation [11]. DHEA is subsequently converted to androstenediol, and androstenedione is transformed into testosterone by the enzymes 17β-hydroxysteroid dehydrogenase (17βHSD) and 3β-hydroxysteroid dehydrogenase (3βHSD). Androstenediol and androstenedione are converted into testosterone by the enzymes 3βHSD and 17β-HSD. In addition, testosterone is transformed into DHT via the enzyme 5α-reductase [12]. Finally, testosterone and androstenedione can be transformed into estrogens by the enzyme aromatase. DHT is the only androgen that is not converted to estrogen. Androstenedione is transformed to estrone by the P450 aromatase and estrone to 17β-estradiol by the enzyme 17β-HSD. Testosterone can also be transformed into 17β-estradiol by P450aromatase [12]. Androgens testosterone and DHT possess a 17β-hydroxyl and a 3-oxo group, and the former and reduction of the latter result in the loss of biological activity [13] (Figure 1).
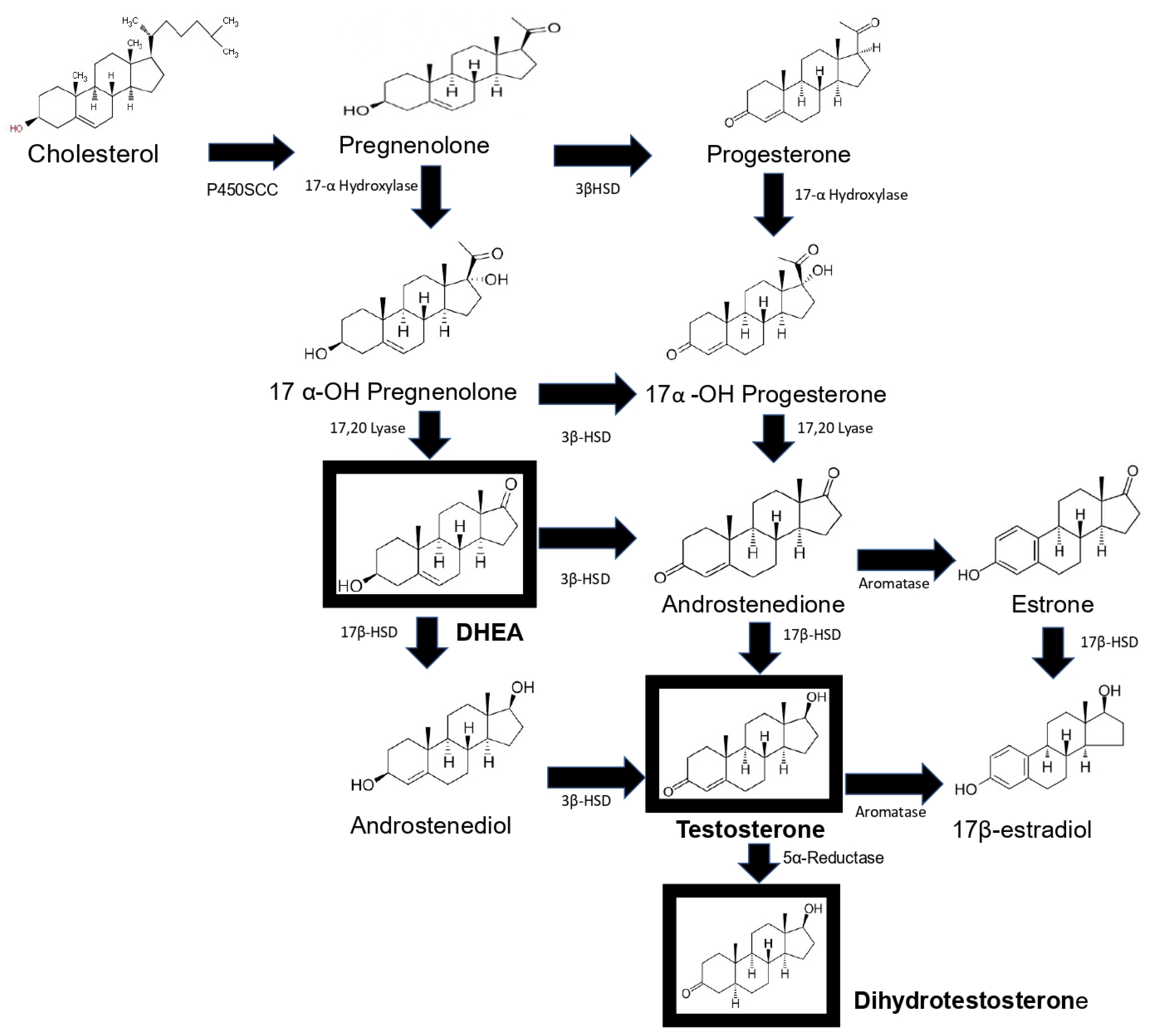
Figure 1. Schematic representation of androgen synthesis. Steroidogenesis starts with the cleavage of cholesterol by the P450SCS enzyme, which transforms it into pregnenolone, which is hydroxylated at carbon 17 by 17α-hydroxylase; the resulting product is the hormone DHEA. DHEA is converted to androstenediol by the enzyme 17β-HSD. This steroid and androstenedione are transformed to testosterone by the enzymes 3β-HSD and 17β-HSD, respectively. Dihydrotestosterone (DHT) is synthesized from testosterone. Finally, androstenedione is converted into estrone by the enzyme aromatase and estrone into 17β-estradiol by 17βHSD, and testosterone is transformed into 17β-estradiol by aromatase. The main sex hormones are highlighted in a box. This image is based on the text described in [12].
Androgens not only determine biological sex but also impact health and disease. The four androgens DHT, testosterone, androstenedione and DHEA act on many cells, including those of the immune system, and influence their function, maturation and susceptibility to damage by autoimmune processes [14]. The biological function of these steroids depends on their concentration, affinity and availability to interact with their receptors [15]. In addition, its concentration varies with sex and declines with age [6]. The potencies of androgens fluctuate; DHT is the most potent (300%), with testosterone (100%), androstenedione (10%) and DHEA (only 5%) [16]. In diseases such as rheumatoid arthritis, systemic lupus erythematosus, or multiple sclerosis, there is a marked sexual dimorphism; women are more susceptible than men. In contrast, in cancer, this pattern is reversed.
Testosterone is the most concentrated androgen in adult men, and DHT constitutes only 10% of the testosterone concentration. In men, 2% of testosterone is free, and 30% binds to sex hormone binding globulin (SHBG) with high affinity; the remaining testosterone binds with lower affinity to albumin and other proteins [17]. Interestingly, only free testosterone has biological activity [18]; therefore, the proteins that bind androgens modulate their action. On the other hand, DHEA binds with low affinity to the AR compared to testosterone or DHT. DHEA-S has high affinity for albumin and a long half-life, and it does not bind to AR [12]. DHEA also binds to estrogen receptors α and β (ERα and ERβ) [19]. Moreover, DHEA and DHEA-S act as ligands for G protein-coupled receptors and several nuclear receptors. Both steroids can modulate different signaling pathways [20]. The above explains the versatility and specificity of the functions that androgens play in the body.
3. Mechanisms of Action of AR-Dependent Androgens (Canonical Pathway)
AR is a single-chain molecule present in the cytoplasm and is a ligand-dependent transcription factor. This receptor has three domains, the N-terminal domain (NTD), the DNA-binding domain (DBD) and the ligand-binding domain (LBD), which are all highly conserved (Figure 2) [21]. For steroid receptors to acquire the proper conformation that allows them to bind to their ligand, a highly ordered maturation process involving chaperones and cochaperones is required [22]. Some of the proteins involved include heat shock protein 90 (Hsp90), a 23 kDa chaperone and an FKBP52 protein containing a peptide with an Hsp90-binding TPR tetratricopeptide domain [23]. This complex is a positive regulator of the AR and for the glucocorticoid receptor GR [24]; the complex dissociates once the receptors have bound to its ligands (testosterone, DHT, androstenedione and DHEA) [25].
The AR–ligand complex dimerizes with another AR–ligand complex, undergoes phosphorylation and translocates to the nucleus, where it binds to androgen response elements (AREs) containing a 15-base pair palindromic sequence; this interaction modulates the expression of multiple androgen-dependent genes [26] (Figure 2). This is important because the action of testosterone and DHEA is mediated by AR, and this receptor is expressed in 80% of breast cancers and has been associated with reduced mortality [27]. It could therefore be used as a prognostic for the disease and as a probable target for therapy.
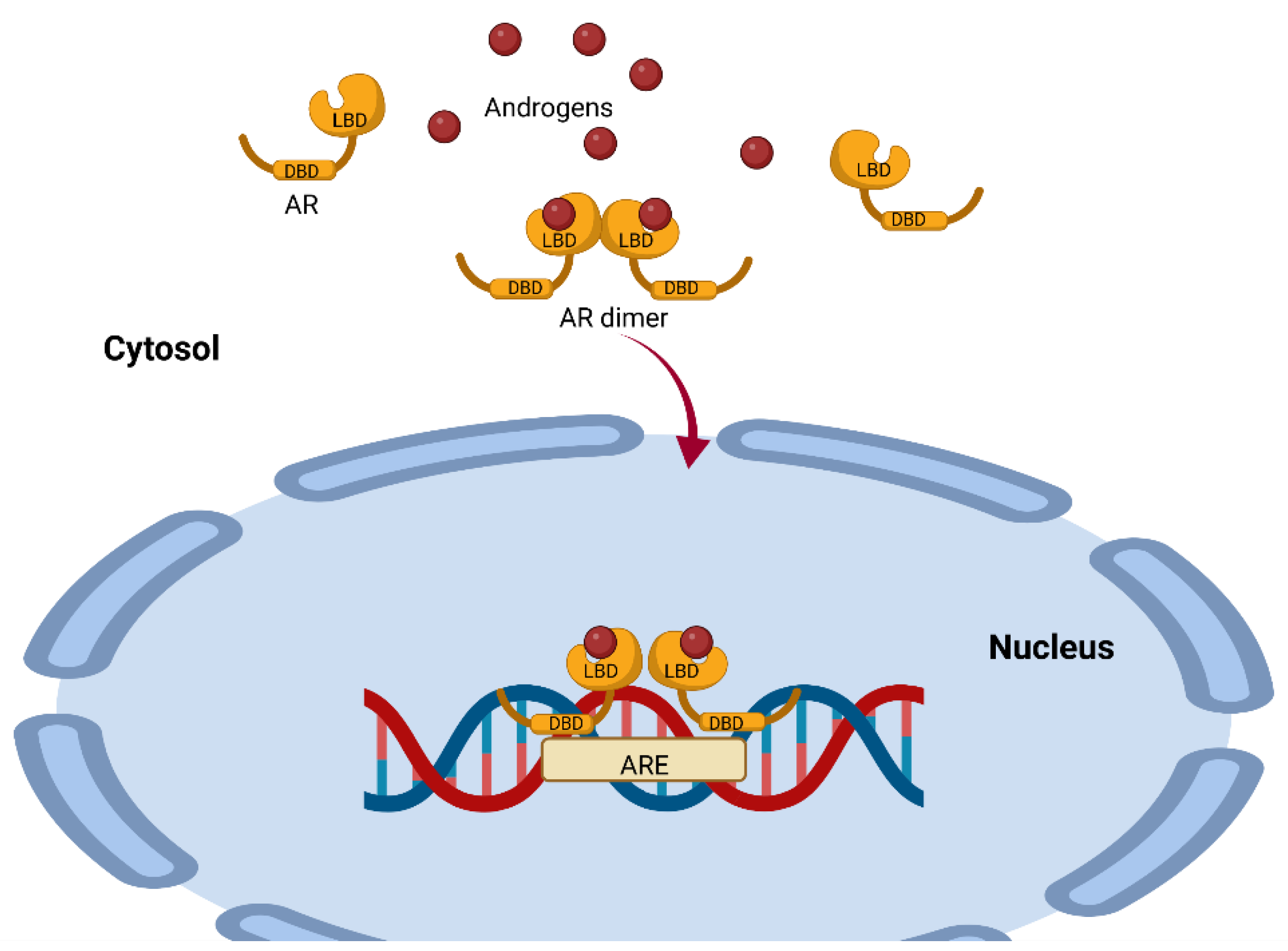
Figure 2. Representation of the mechanism of androgen action mediated by the androgen receptor. The AR has a highly conserved ligand-binding region (LBD) and a highly conserved DNA-binding region (DBD). Once the ligand binds to the AR, it dimerizes with another AR–ligand complex and translocates to the nucleus, where it binds to androgen response elements (AREs), modulating the expression of several genes. This image was based on the text of [28] and created using the BioRender.com software.
The number of ARs is higher in males than in females and depends on the concentration of androgens [6]. The variety of androgens and the different affinities for AR are important for understanding the regulation of the immune response since AR is found in macrophages, T lymphocytes, B cells, neutrophils, dendritic cells and NK cells [29].
4. AR-Independent Androgen Mechanisms of Action (Noncanonical Pathway)
Androgens also act independently of the binding of AR to DNA through noncanonical pathways, which include alternative nongenomic signaling pathways triggered by androgens via G protein-coupled receptors (GPCRs) or binding to the androgen receptor in membranes such as ZIP9, which, by activating G proteins and second messengers, increases intracellular zinc and the expression of proapoptotic genes, leading to apoptosis [30][31]. Androgen-independent signaling can also occur, which ultimately impacts androgen signaling pathways or includes an integration of nongenomic and genomic responses, as in the case of protein kinase A activation [32], or calcium signaling can occur; in fact, testosterone induces the release of intracellular calcium [33].
Noncanonical pathways are characterized by the fact that they occur within minutes compared to the canonical pathway, which lasts for hours; it starts when androgens interact with receptors present on the plasma membrane (mAR) and affect cytosolic signaling pathways such as Src/Ras/Raf/Erk1/2 [34]; these pathways are activated by growth factors such as Src/Akt/PI3K [35]. In addition, some membrane receptors that bind testosterone are G protein-coupled receptors, and this interaction phosphorylates ErK1/2, CREB, and ATF-1 [36]. Another androgen-associated membrane receptor is TRPM8, which acts as a calcium-permeable channel [37]. Overexpression of TRPM8 decreases the cytotoxic activity of CD8+ T cells, which facilitates the proliferation of cancer cells [38]. Additionally, the membrane receptor GPRC6A translates the effects of androgens, and overexpression of this receptor allows HEK-293 cells lacking AR to respond to testosterone [39]. In addition, AR mutants unable to bind DNA after incubation with DHT increase the activity of protein kinase B (Akt), ERK kinases and mitogen-activated protein kinases (MAPKs) [40]. Moreover, the ERK/MAPK pathway is important because it increases the IL-2 and IFN-γ concentrations of CD8+ cells [41] and the expression of IL-4 and IL-10 in lymphocytes [42]. Finally, the membrane receptor GPRC in macrophages binds androgens and activates PI3K kinase, which phosphorylates inositol 3,4 diphosphate (PIP2), transforming it into inositol 3,4,5 triphosphate (PIP3). This induces the activation of Akt kinase, which in turn inhibits TSC1/2, allowing the activity of MTORC1 kinase, which phosphorylates P70S6K kinase. This enhances the expression of IL1β, IL-6 and TNF-α in macrophages [43] (Figure 3). The above is important because these cytokines are increased in response to bacterial and viral infections [44][45]. Knowledge of the mechanisms of androgen action on their target cells will contribute to understanding the relationship of androgens to susceptibility to infectious diseases, autoimmune disorders, and cancer.
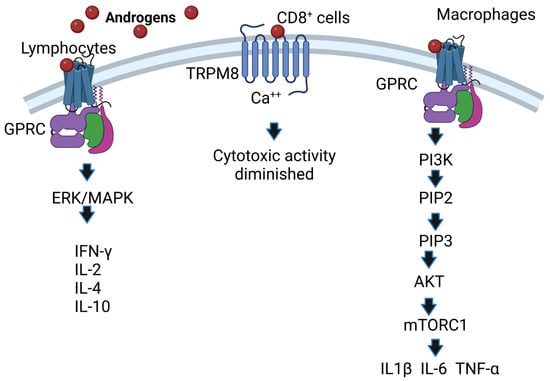
Figure 3. AR-independent effects of androgens. Androgens bind to membrane receptors that trigger the activation of the ERK (MAPK) pathway in lymphocytes, and IFN-γ, IL-2, IL-4 and IL-10 expression is increased. Likewise, androgens activate the phosphatidylinositol-3 kinase (PI3K) pathway; if this pathway is activated in macrophages, it regulates the expression of the cytokines IL1β, IL-6 and TNF-α. Image created based on the concepts described in [43] using the BioRender.com software. The interaction of androgens with TRPM8 causes Ca++ to increase, which decreases the activity of cytotoxic T cells.
5. Effect of Androgens on Lymphoid Organs
Several immune response cells proliferate in the spleen or mature in the thymus, and androgens regulate cell proliferation in both organs. Gonadectomy regenerates thymus size in aged male rats and increases lymphocyte proliferation, which is reversed by testosterone administration [46]. In contrast, the use of the aromatase inhibitor ATD regenerates thymus size, demonstrating the importance of testosterone biotransformation in this phenomenon [47]. In addition, testosterone promotes local glucocorticoid synthesis and release, which causes thymus involution by increasing apoptosis in the thymus tissue, and mice lacking the glucocorticoid receptor decrease thymus size [48]. This finding is important because the thymus is the organ in which bone marrow-derived T-cell precursors mature and would explain at least in part the immunosuppressive activity of testosterone in various parasitic diseases. For example, in Trypanosoma cruzi infections, testosterone administration causes thymus atrophy by promoting apoptosis through increased TNF-α. In contrast, DHEA administration induces thymocyte proliferation and reduces TNF-α concentration; low levels of this cytokine confer protection against this parasite. However, combined testosterone and DHEA treatment improves the immune response by reducing the number of parasites and the suppressive effects of testosterone, and by lowering TNF-α levels [3]. Furthermore, it has been described that DHEA counteracts glucocorticoid-induced thymic involution in vitro and in vivo [49]. In contrast, DHEA reduces thymus size in healthy female rats [50], confirming that the effect of DHEA is sex-dependent. Regarding other androgens, DHT increases the expression of autoimmune regulatory transcription factor (AIRE) in the thymus [8], which promotes immune tolerance and leads to a lower incidence of autoimmune diseases. DHT also decreases splenocyte proliferation in vivo [51]. In contrast, DHEA reverses the effects of corticosteroids and enhances T-lymphocyte functions by downregulating proinflammatory cytokines such as TNF-α.
6. Effect of Androgens on Toll-Like Receptors
Toll-like receptors (TLRs) are recognition systems relevant to the innate immune response, as they detect damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) [52]. Androgens regulate the expression of these receptors; DHEA increases the expression of TLR2 and TLR4 in macrophages of mice with sepsis, both receptors censor Gram-positive and Gram-negative bacteria, respectively [53]. However, it is not known whether DHEA modulates the expression of these TLRs in a straightforward manner. However, DHEA improves phagocytosis but does not affect TLR4 expression in neutrophils in a Salmonella enterica model [54]. One possible mechanism involved is that DHEA modulates the alternative splicing of TLR4, which has several spliceosomes [55], because DHEA modulates the expression of molecules such as the β-glucocorticoid receptor by this mechanism [56]. Furthermore, testosterone decreases TLR4 expression in macrophages stimulated in vitro and in lipopolysaccharide-stimulated male mice. In addition, orchectomy increases TLR4 expression [57]. In addition, the increased TLR4 expression correlates with increased AR expression in a hepatocarcinoma cell model where silencing AR with siRNA decreases the DHT-dependent increase in TLR4 [58]. In addition, testosterone increases TLR6 expression associated with the epigenetic modification caused by TLR6 promoter methylation and decreases TLR8 expression in the liver of Plasmodium chabaudi-infected mice, contributing to the persistence of infection [59]. On the other hand, DHT further increases TLR7 and inhibits TLR9 expression on dendritic cells by inhibiting endothelial cell apoptosis, a mechanism that is associated with the maintenance of immune tolerance [60]. These findings show that androgens suppress the expression of TLR-4, TLR-8, and TLR-9 receptors. In contrast, androgens also increase TLR4, TL-6 and TLR7 expression. These findings suggest that although the main modulatory mechanism of androgens on TLRs is the AR, there must be specific regulatory mechanisms for each cell group, which partially explains the increased susceptibility of males to a variety of infectious diseases.
References
- Benten, W.P.; Ulrich, P.; Kuhn-Velten, W.N.; Vohr, H.W.; Wunderlich, F. Testosterone-induced susceptibility to Plasmodium chabaudi malaria: Persistence after withdrawal of testosterone. J. Endocrinol. 1997, 153, 275–281.
- Galindo-Sevilla, N.; Soto, N.; Mancilla, J.; Cerbulo, A.; Zambrano, E.; Chavira, R.; Huerto, J. Low serum levels of dehydroepiandrosterone and cortisol in human diffuse cutaneous leishmaniasis by Leishmania mexicana. Am. J. Trop. Med. Hyg. 2007, 76, 566–572.
- Filipin Mdel, V.; Caetano, L.C.; Brazao, V.; Santello, F.H.; Toldo, M.P.; do Prado, J.C., Jr. DHEA and testosterone therapies in Trypanosoma cruzi-infected rats are associated with thymic changes. Res. Vet. Sci. 2010, 89, 98–103.
- Vom Steeg, L.G.; Klein, S.L. Sex Steroids Mediate Bidirectional Interactions between Hosts and Microbes. Horm. Behav. 2017, 88, 45–51.
- Klein, S.L.; Flanagan, K.L. Sex differences in immune responses. Nat. Rev. Immunol. 2016, 16, 626–638.
- Lu, S.F.; McKenna, S.E.; Cologer-Clifford, A.; Nau, E.A.; Simon, N.G. Androgen receptor in mouse brain: Sex differences and similarities in autoregulation. Endocrinology 1998, 139, 1594–1601.
- Muniyappa, R.; Wong, K.A.; Baldwin, H.L.; Sorkin, J.D.; Johnson, M.L.; Bhasin, S.; Harman, S.M.; Blackman, M.R. Dehydroepiandrosterone secretion in healthy older men and women: Effects of testosterone and growth hormone administration in older men. J. Clin. Endocrinol. Metab. 2006, 91, 4445–4452.
- Zhu, M.L.; Bakhru, P.; Conley, B.; Nelson, J.S.; Free, M.; Martin, A.; Starmer, J.; Wilson, E.M.; Su, M.A. Sex bias in CNS autoimmune disease mediated by androgen control of autoimmune regulator. Nat. Commun. 2016, 7, 11350.
- Constantopoulos, G.; Tchen, T.T. Cleavage of cholesterol side chain by adrenal cortex. I. Cofactor requirement and product of clevage. J. Biol. Chem. 1961, 236, 65–67.
- Simpson, E.R.; Boyd, G.S. The cholesterol side-chain cleavage system of bovine adrenal cortex. Eur. J. Biochem. 1967, 2, 275–285.
- Adamkiewicz, M.; Zgliczynski, S.; Sfowinska-Srzednicka, J.; Jeske, W.; Rabijewski, M.; Pietrzyk, E.; Srzednicki, M.; Sadowski, Z. The relationship between plasma androgens (dehydroepiandrosterone sulfate and testosterone), insulin, coagulation and fibrinolytic factors in men with coronary arteriosclerosis. Aging Male 1998, 1, 270–279.
- Miller, W.L.; Auchus, R.J. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 2011, 32, 81–151.
- Fragkaki, A.G.; Angelis, Y.S.; Koupparis, M.; Tsantili-Kakoulidou, A.; Kokotos, G.; Georgakopoulos, C. Structural characteristics of anabolic androgenic steroids contributing to binding to the androgen receptor and to their anabolic and androgenic activities. Applied modifications in the steroidal structure. Steroids 2009, 74, 172–197.
- Gubbels Bupp, M.R.; Jorgensen, T.N. Androgen-Induced Immunosuppression. Front. Immunol. 2018, 9, 794.
- Zhou, Z.X.; Lane, M.V.; Kemppainen, J.A.; French, F.S.; Wilson, E.M. Specificity of ligand-dependent androgen receptor stabilization: Receptor domain interactions influence ligand dissociation and receptor stability. Mol. Endocrinol. 1995, 9, 208–218.
- Marchetti, P.M.; Barth, J.H. Clinical biochemistry of dihydrotestosterone. Ann. Clin. Biochem. 2013, 50 Pt 2, 95–107.
- Dunn, J.F.; Nisula, B.C.; Rodbard, D. Transport of steroid hormones: Binding of 21 endogenous steroids to both testosterone-binding globulin and corticosteroid-binding globulin in human plasma. J. Clin. Endocrinol. Metab. 1981, 53, 58–68.
- Morales, A.; Buvat, J.; Gooren, L.J.; Guay, A.T.; Kaufman, J.M.; Tan, H.M.; Torres, L.O. Endocrine aspects of sexual dysfunction in men. J. Sex. Med. 2004, 1, 69–81.
- Miller, K.K.; Al-Rayyan, N.; Ivanova, M.M.; Mattingly, K.A.; Ripp, S.L.; Klinge, C.M.; Prough, R.A. DHEA metabolites activate estrogen receptors alpha and beta. Steroids 2013, 78, 15–25.
- Prough, R.A.; Clark, B.J.; Klinge, C.M. Novel mechanisms for DHEA action. J. Mol. Endocrinol. 2016, 56, R139–R155.
- Tan, M.H.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E.L. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 2015, 36, 3–23.
- Smith, D.F.; Toft, D.O. Minireview: The intersection of steroid receptors with molecular chaperones: Observations and questions. Mol. Endocrinol. 2008, 22, 2229–2240.
- De Leon, J.T.; Iwai, A.; Feau, C.; Garcia, Y.; Balsiger, H.A.; Storer, C.L.; Suro, R.M.; Garza, K.M.; Lee, S.; Sang Kim, Y. Targeting the regulation of androgen receptor signaling by the heat shock protein 90 cochaperone FKBP52 in prostate cancer cells. Proc. Natl. Acad. Sci. USA 2011, 108, 11878–11883.
- Riggs, D.L.; Roberts, P.J.; Chirillo, S.C.; Cheung-Flynn, J.; Prapapanich, V.; Ratajczak, T.; Gaber, R.; Picard, D.; Smith, D.F. The Hsp90-binding peptidylprolyl isomerase FKBP52 potentiates glucocorticoid signaling in vivo. EMBO J. 2003, 22, 1158–1167.
- Chang, C.; Saltzman, A.; Yeh, S.; Young, W.; Keller, E.; Lee, H.J.; Wang, C.; Mizokami, A. Androgen receptor: An overview. Crit. Rev. Eukaryot. Gene Expr. 1995, 5, 97–125.
- Massie, C.E.; Adryan, B.; Barbosa-Morais, N.L.; Lynch, A.G.; Tran, M.G.; Neal, D.E.; Mills, I.G. New androgen receptor genomic targets show an interaction with the ETS1 transcription factor. EMBO Rep. 2007, 8, 871–878.
- Hu, R.; Dawood, S.; Holmes, M.D.; Collins, L.C.; Schnitt, S.J.; Cole, K.; Marotti, J.D.; Hankinson, S.E.; Colditz, G.A.; Tamimi, R.M. Androgen Receptor Expression and Breast Cancer Survival in Postmenopausal WomenAndrogen Receptor Expression and Breast Cancer Survival. Clin. Cancer Res. 2011, 17, 1867–1874.
- Davey, R.A.; Grossmann, M. Androgen Receptor Structure, Function and Biology: From Bench to Bedside. Clin. Biochem. Rev. 2016, 37, 3–15.
- Lai, J.J.; Lai, K.P.; Zeng, W.; Chuang, K.H.; Altuwaijri, S.; Chang, C. Androgen receptor influences on body defense system via modulation of innate and adaptive immune systems: Lessons from conditional AR knockout mice. Am. J. Pathol. 2012, 181, 1504–1512.
- Xia, C.; Ma, W.; Wang, F.; Hua, S.; Liu, M. Identification of a prostate-specific G-protein coupled receptor in prostate cancer. Oncogene 2001, 20, 5903–5907.
- Thomas, P.; Pang, Y.; Dong, J. Membrane androgen receptor characteristics of human ZIP9 (SLC39A) zinc transporter in prostate cancer cells: Androgen-specific activation and involvement of an inhibitory G protein in zinc and MAP kinase signaling. Mol. Cell. Endocrinol. 2017, 447, 23–34.
- Nazareth, L.V.; Weigel, N.L. Activation of the human androgen receptor through a protein kinase A signaling pathway. J. Biol. Chem. 1996, 271, 19900–19907.
- Estrada, M.; Espinosa, A.; Muller, M.; Jaimovich, E. Testosterone stimulates intracellular calcium release and mitogen-activated protein kinases via a G protein-coupled receptor in skeletal muscle cells. Endocrinology 2003, 144, 3586–3597.
- Falkenstein, E.; Tillmann, H.C.; Christ, M.; Feuring, M.; Wehling, M. Multiple actions of steroid hormones—A focus on rapid, nongenomic effects. Pharmacol. Rev. 2000, 52, 513–556.
- Valverde, M.A.; Parker, M.G. Classical and novel steroid actions: A unified but complex view. Trends Biochem. Sci. 2002, 27, 172–173.
- Shihan, M.; Bulldan, A.; Scheiner-Bobis, G. Non-classical testosterone signaling is mediated by a G-protein-coupled receptor interacting with Gnalpha11. Biochim. Biophys. Acta 2014, 1843, 1172–1181.
- Zhang, L.; Barritt, G.J. Evidence that TRPM8 is an androgen-dependent Ca2+ channel required for the survival of prostate cancer cells. Cancer Res. 2004, 64, 8365–8373.
- Lan, X.; Zhao, J.; Song, C.; Yuan, Q.; Liu, X. TRPM8 facilitates proliferation and immune evasion of esophageal cancer cells. Biosci. Rep. 2019, 39, BSR20191878.
- Pi, M.; Parrill, A.L.; Quarles, L.D. GPRC6A mediates the non-genomic effects of steroids. J. Biol. Chem. 2010, 285, 39953–39964.
- Kang, H.Y.; Cho, C.L.; Huang, K.L.; Wang, J.C.; Hu, Y.C.; Lin, H.K.; Chang, C.; Huang, K.E. Nongenomic androgen activation of phosphatidylinositol 3-kinase/Akt signaling pathway in MC3T3-E1 osteoblasts. J. Bone Miner. Res. Off. J. Am. Soc. Bone Mineral. Res. 2004, 19, 1181–1190.
- Damasio, M.P.; Marchingo, J.M.; Spinelli, L.; Hukelmann, J.L.; Cantrell, D.A.; Howden, A.J.M. Extracellular signal-regulated kinase (ERK) pathway control of CD8+ T cell differentiation. Biochem. J. 2021, 478, 79–98.
- Song, G.Y.; Chung, C.S.; Chaudry, I.H.; Ayala, A. MAPK p38 antagonism as a novel method of inhibiting lymphoid immune suppression in polymicrobial sepsis. Am. J. Physiol. Cell Physiol. 2001, 281, C662–C669.
- Xie, S.; Chen, M.; Yan, B.; He, X.; Chen, X.; Li, D. Identification of a role for the PI3K/AKT/mTOR signaling pathway in innate immune cells. PLoS ONE 2014, 9, e94496.
- Carvalho, N.B.; de Lourdes Bastos, M.; Souza, A.S.; Netto, E.M.; Arruda, S.; Santos, S.B.; Carvalho, E.M. Impaired TNF, IL-1beta, and IL-17 production and increased susceptibility to Mycobacterium tuberculosis infection in HTLV-1 infected individuals. Tuberculosis 2018, 108, 35–40.
- Matsuda, K.; Tsutsumi, H.; Sone, S.; Yoto, Y.; Oya, K.; Okamoto, Y.; Ogra, P.L.; Chiba, S. Characteristics of IL-6 and TNF-alpha production by respiratory syncytial virus-infected macrophages in the neonate. J. Med. Virol. 1996, 48, 199–203.
- Olsen, N.J.; Kovacs, W.J. Effects of androgens on T and B lymphocyte development. Immunol. Res. 2001, 23, 281–288.
- Greenstein, B.D.; de Bridges, E.F.; Fitzpatrick, F.T. Aromatase inhibitors regenerate the thymus in aging male rats. Int. J. Immunopharmacol. 1992, 14, 541–553.
- Chen, Y.; Qiao, S.; Tuckermann, J.; Okret, S.; Jondal, M. Thymus-derived glucocorticoids mediate androgen effects on thymocyte homeostasis. FASEB J. 2010, 24, 5043–5051.
- May, M.; Holmes, E.; Rogers, W.; Poth, M. Protection from glucocorticoid induced thymic involution by dehydroepiandrosterone. Life Sci. 1990, 46, 1627–1631.
- Parker, C.R., Jr.; Conway-Myers, B.A. The effects of dehydroepiandrosterone (DHEA) on the thymus, spleen, and adrenals of prepubertal and adult female rats. Endocr. Res. 1998, 24, 113–126.
- Angele, M.K.; Ayala, A.; Cioffi, W.G.; Bland, K.I.; Chaudry, I.H. Testosterone: The culprit for producing splenocyte immune depression after trauma hemorrhage. Am. J. Physiol. 1998, 274, C1530–C1536.
- Naqvi, I.; Giroux, N.; Olson, L.; Morrison, S.A.; Llanga, T.; Akinade, T.O.; Zhu, Y.; Zhong, Y.; Bose, S.; Arvai, S.; et al. DAMPs/PAMPs induce monocytic TLR activation and tolerance in COVID-19 patients; nucleic acid binding scavengers can counteract such TLR agonists. Biomaterials 2022, 283, 121393.
- Matsuda, A.; Furukawa, K.; Suzuki, H.; Matsutani, T.; Tajiri, T.; Chaudry, I.H. Dehydroepiandrosterone modulates toll-like receptor expression on splenic macrophages of mice after severe polymicrobial sepsis. Shock 2005, 24, 364–369.
- Brauer, V.S.; Zambuzi, F.A.; Espíndola, M.S.; Cavalcanti Neto, M.P.; Prado, M.K.B.; Cardoso, P.M.; Soares, L.S.; Galvao-Lima, L.J.; Leopoldino, A.M.; Cardoso, C.R.d.B. The influence of dehydroepiandrosterone on effector functions of neutrophils. Braz. J. Pharm. Sci. 2021, 57, 419139.
- Wang, X.; Pei, J.; Bao, P.; Liang, C.; Chu, M.; Guo, S.; Yan, P.; Guo, X. Identification of Yak’s TLR4 Alternative Spliceosomes and Bioinformatic Analysis of TLR4 Protein Structure and Function. Animals 2020, 11, 32.
- Buoso, E.; Galasso, M.; Ronfani, M.; Serafini, M.M.; Lanni, C.; Corsini, E.; Racchi, M. Role of spliceosome proteins in the regulation of glucocorticoid receptor isoforms by cortisol and dehydroepiandrosterone. Pharmacol. Res. 2017, 120, 180–187.
- Rettew, J.A.; Huet-Hudson, Y.M.; Marriott, I. Testosterone reduces macrophage expression in the mouse of toll-like receptor 4, a trigger for inflammation and innate immunity. Biol. Reprod. 2008, 78, 432–437.
- Han, Q.; Yang, D.; Yin, C.; Zhang, J. Androgen Receptor (AR)-TLR4 Crosstalk Mediates Gender Disparities in Hepatocellular Carcinoma Incidence and Progression. J. Cancer 2020, 11, 1094–1103.
- Al-Quraishy, S.; Dkhil, M.A.; Abdel-Baki, A.A.; Arauzo-Bravo, M.J.; Delic, D.; Wunderlich, F. Testosterone persistently dysregulates hepatic expression of Tlr6 and Tlr8 induced by Plasmodium chabaudi malaria. Parasitol. Res. 2014, 113, 3609–3620.
- Ainola, M.; Porola, P.; Takakubo, Y.; Przybyla, B.; Kouri, V.P.; Tolvanen, T.A.; Hanninen, A.; Nordstrom, D.C. Activation of plasmacytoid dendritic cells by apoptotic particles-mechanism for the loss of immunological tolerance in Sjogren’s syndrome. Clin. Exp. Immunol. 2018, 191, 301–310.
More
Information
Subjects:
Endocrinology & Metabolism
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.4K
Revisions:
2 times
(View History)
Update Date:
15 Dec 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No