 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Andrey Buglak | -- | 3431 | 2022-12-12 13:12:29 | | | |
| 2 | Amina Yu | -54 word(s) | 3377 | 2022-12-13 02:18:16 | | | | |
| 3 | Amina Yu | -2 word(s) | 3375 | 2022-12-13 02:19:24 | | |
Video Upload Options
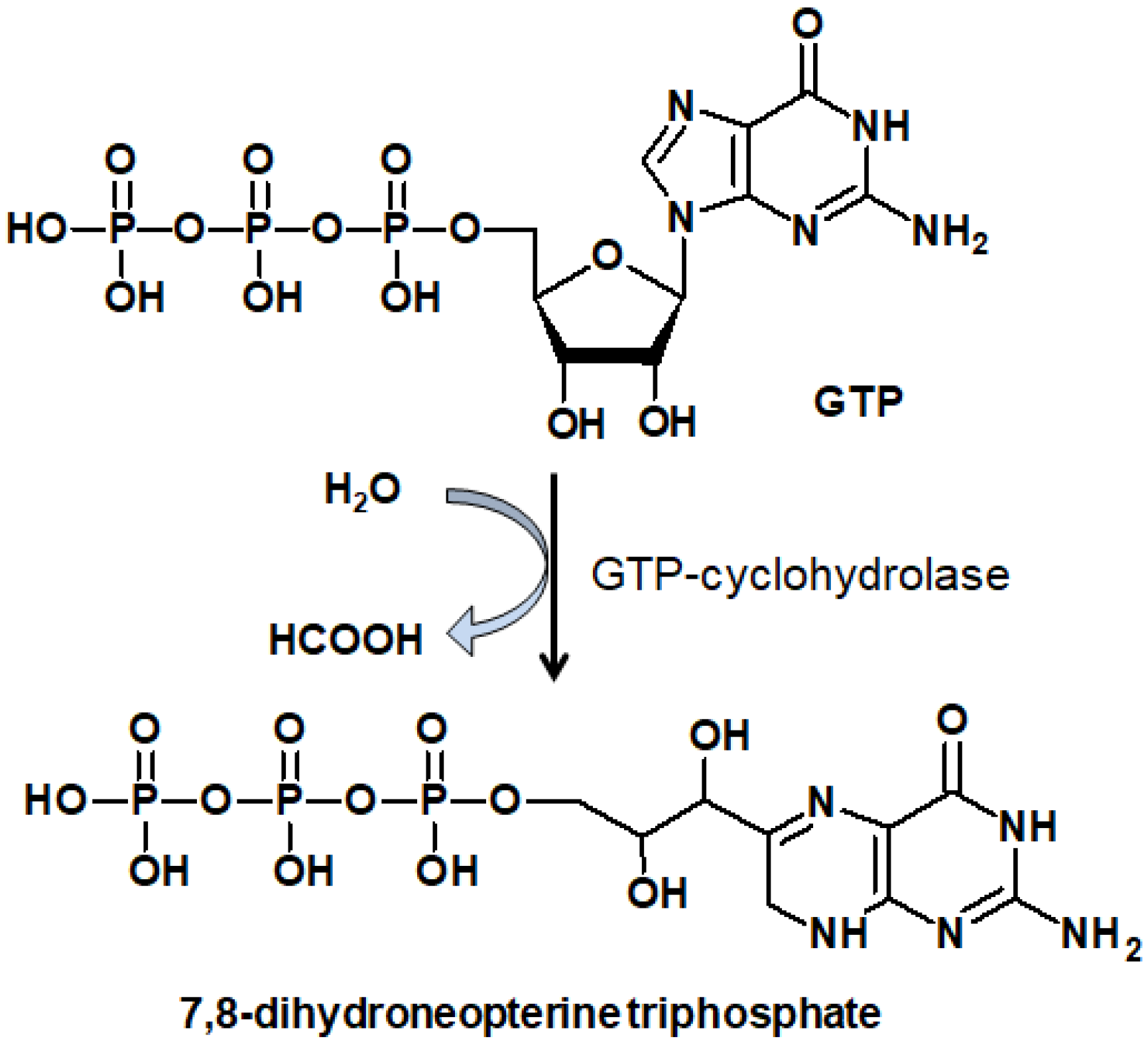
Pterins are low-molecular weight heterocyclic compounds widely distributed in living organisms, primarily as reduced coenzymes. Structurally, pterins are a conjugated system of pyrazine and pyrimidine rings, the so-called pteridine, which is accompanied by a carbonyl group (C=O) at the C4 position and an amino group (NH2) at the C2 position. The pteridine structure is also characteristic of folates (folic acid and its derivatives) and flavins, or benzopteridines, which are derivatives of isoalloxazine. Folates are usually called “conjugated pterins” since they possess a para-aminobenzoilglutamine residue, whereas pterins are called “unconjugated pterins”. In addition to pterin, folates include a para-aminobenzoic acid (p-ABA) residue and one to five glutamic acid (Glu) residues.
1. Evidence of Pteridine Participation in Photoreception
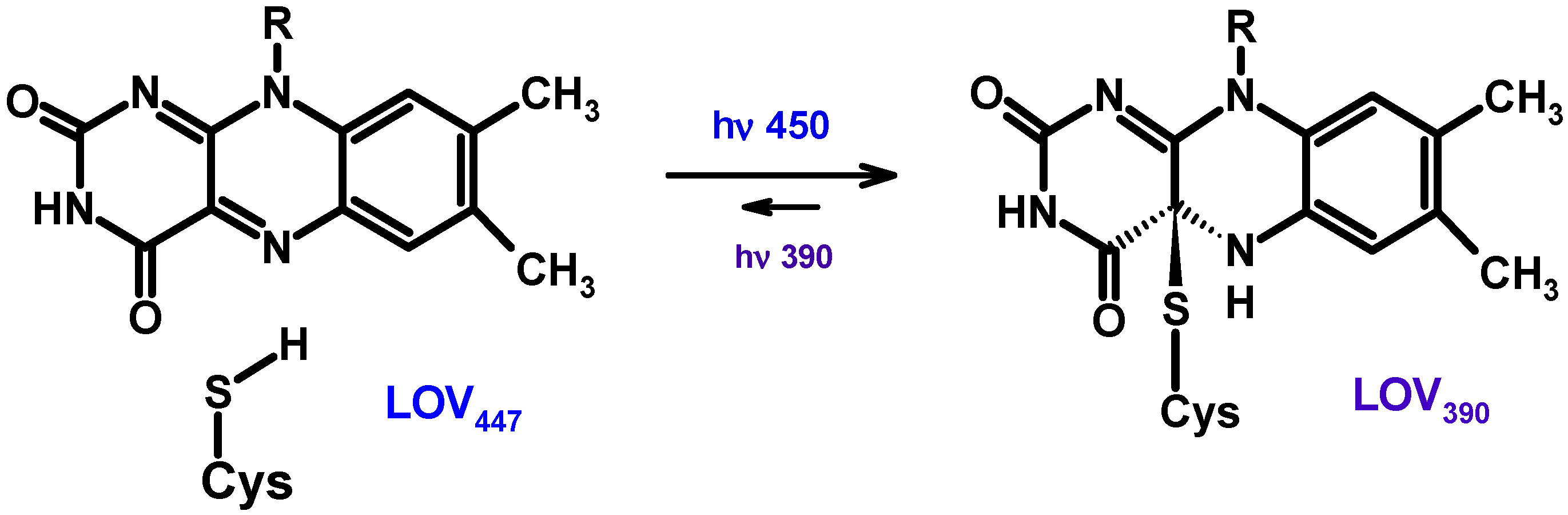
- ➢
- ➢
-
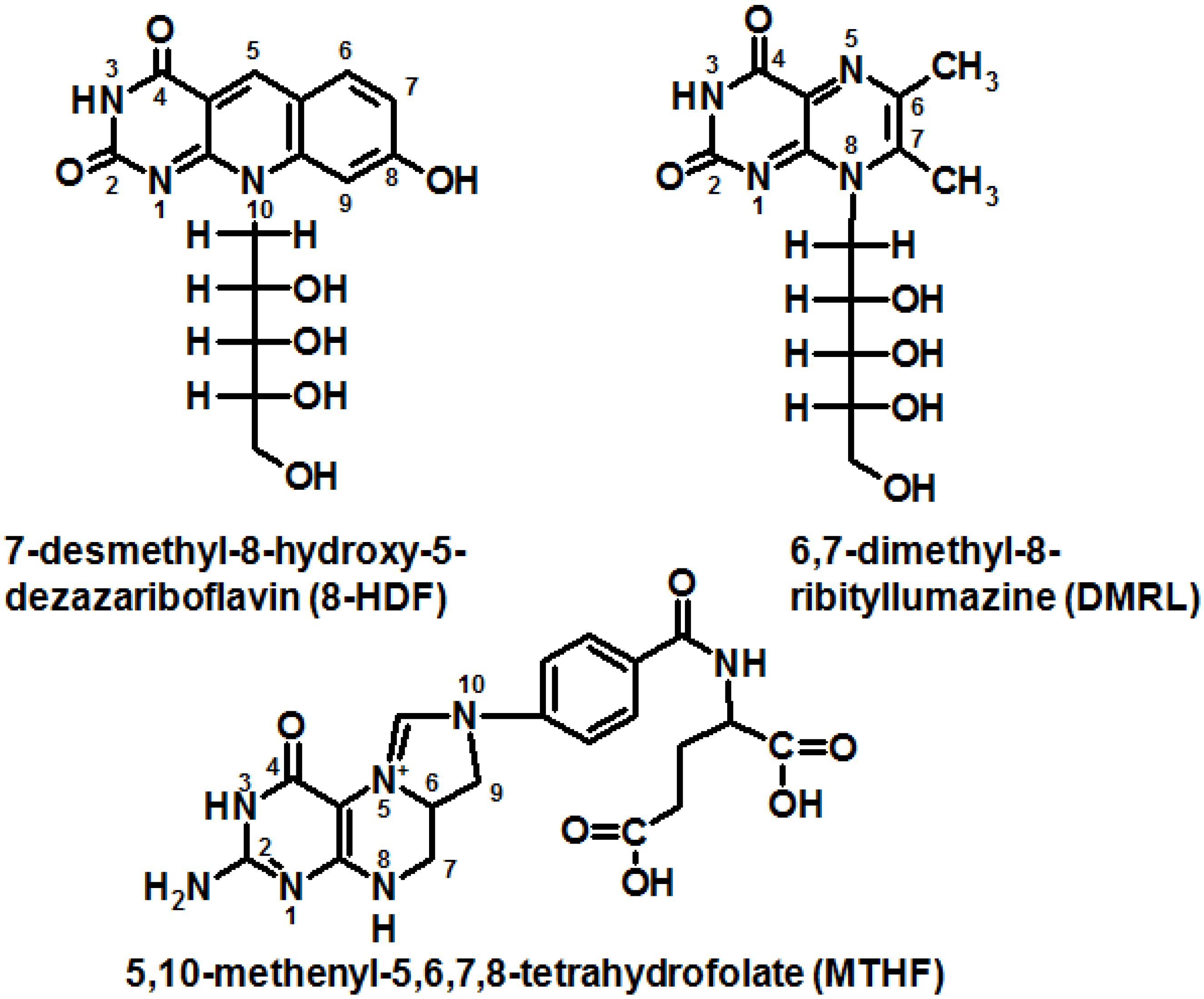
7-desmethyl-8-hydroxy-5-deazariboflavin (8-HDF)—in some prokaryotes and protozoa eukaryotes, which have a biosynthetic pathway for this compound [27];
- ➢
-
FMN or the second FAD can also function as an antenna [28];
- ➢
-
it has recently been shown that 6,7-dimethyl-8-ribityllumazine can function as an antenna in some prokaryotic (6-4)-photolyases (Figure 3) [29].
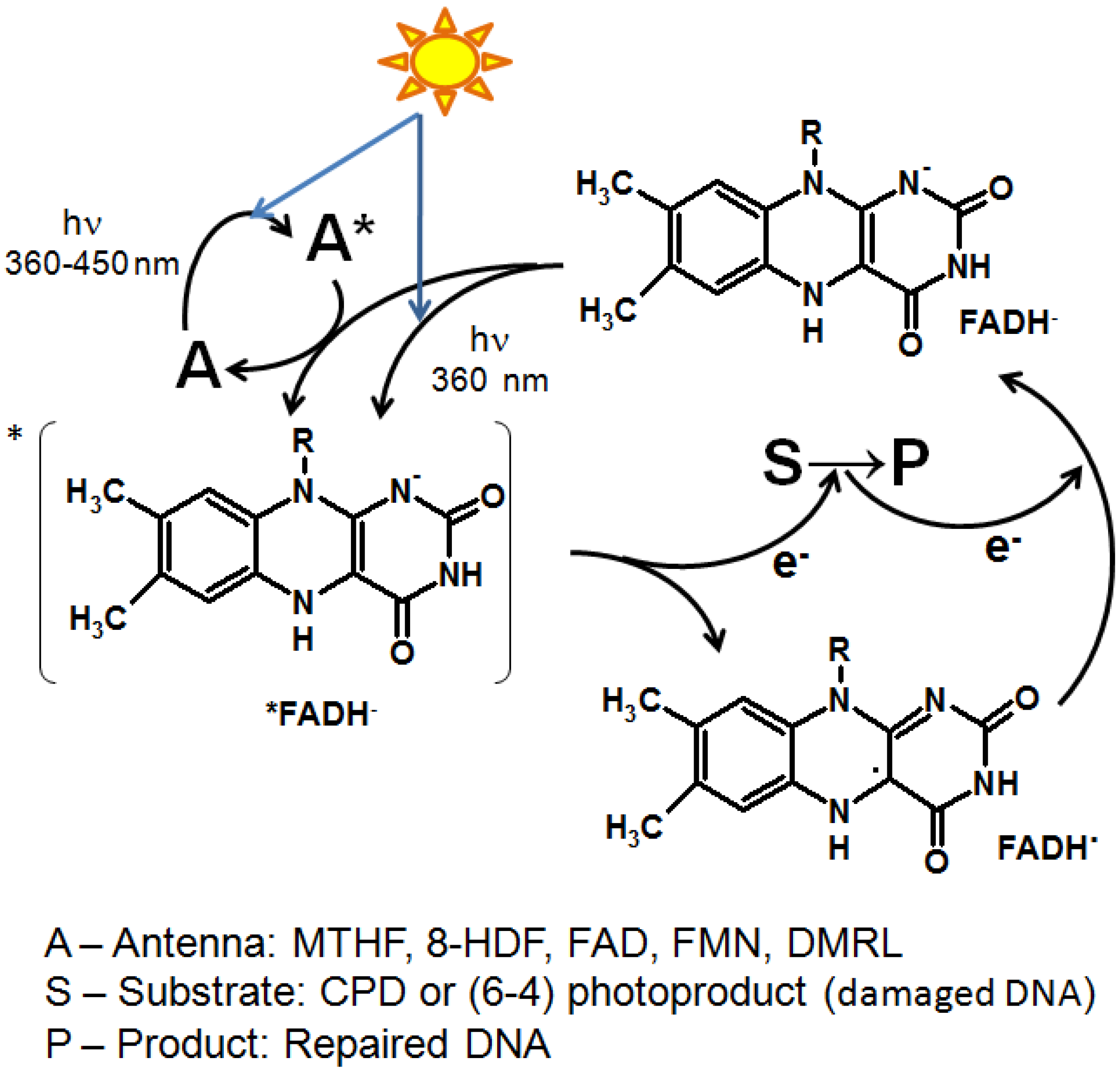
2. The Role of Pterin Coenzymes in the Photoregulation of Metabolism
3. Evolutionary Aspects of Pterin Photochemistry
References
- Kritsky, M.S.; Telegina, T.A.; Vechtomova, Y.L.; Buglak, A.A. Why flavins are not competitors of chlorophyll in the evolution of biological converters of solar energy. Int. J. Mol. Sci. 2013, 14, 575.
- Kavakli, I.H.; Baris, I.; Tardu, M.; Gül, Ş.; Öner, H.; Çal, S.; Bulut, S.; Yarparvar, D.; Berkel, Ç.; Ustaoğlu, P.; et al. The Photolyase/Cryptochrome Family of Proteins as DNA Repair Enzymes and Transcriptional Repressors. Photochem. Photobiol. 2017, 93, 93–103.
- Hosokawa, Y.; Müller, P.; Kitoh-Nishioka, H.; Iwai, S.; Yamamoto, J. Limited solvation of an electron donating tryptophan stabilizes a photoinduced charge-separated state in plant (6–4) photolyase. Sci. Rep. 2022, 12, 5084.
- Cellini, A.; Shankar, M.K.; Wahlgren, W.Y.; Nimmrich, A.; Furrer, A.; James, D.; Wranik, M.; Aumonier, S.; Beale, E.V.; Dworkowski, F.; et al. Structural basis of the radical pair state in photolyases and cryptochromes. Chem. Commun. 2022, 58, 4889–4892.
- Losi, A.; Mandalari, C.; Gärtner, W. From Plant Infectivity to Growth Patterns: The Role of Blue-Light Sensing in the Prokaryotic World. Plants 2014, 3, 70–94.
- Park, S.-Y.; Tame, J.R.H. Seeing the light with BLUF proteins. Biophys. Rev. 2017, 9, 169–176.
- Gauden, M.; Yeremenko, S.; Laan, W.; van Stokkum, I.H.M.; Ihalainen, J.A.; van Grondelle, R.; Hellingwerf, K.J.; Kennis, J.T.M. Photocycle of the Flavin-Binding Photoreceptor AppA, a Bacterial Transcriptional Antirepressor of Photosynthesis Genes. Biochemistry 2005, 44, 3653–3662.
- Goings, J.J.; Li, P.; Zhu, Q.; Hammes-Schiffer, S. Formation of an unusual glutamine tautomer in a blue light using flavin photocycle characterizes the light-adapted state. Proc. Natl. Acad. Sci. USA 2020, 117, 26626–26632.
- Tokonami, S.; Onose, M.; Nakasone, Y.; Terazima, M. Slow Conformational Changes of Blue Light Sensor BLUF Proteins in Milliseconds. J. Am. Chem. Soc. 2022, 144, 4080–4090.
- Shibata, K.; Nakasone, Y.; Terazima, M. Selective Photoinduced Dimerization and Slow Recovery of a BLUF Domain of EB1. J. Phys. Chem. B 2022, 126, 1024–1033.
- Kang, X.-W.; Chen, Z.; Zhou, Z.; Zhou, Y.; Tang, S.; Zhang, Y.; Zhang, T.; Ding, B.; Zhong, D. Direct Observation of Ultrafast Proton Rocking in the BLUF Domain. Angew. Chem. Int. Ed. 2022, 61, e202114423.
- Sancar, A. Mechanisms of DNA Repair by Photolyase and Excision Nuclease (Nobel Lecture). Angew. Chem. Int. Ed. 2016, 55, 8502–8527.
- Kavakli, I.H.; Ozturk, N.; Gul, S. DNA repair by photolyases. Adv. Protein Chem. Struct. Biol. 2019, 115, 1–19.
- Vechtomova, Y.L.; Telegina, T.A.; Kritsky, M.S. Evolution of Proteins of the DNA Photolyase/Cryptochrome Family. Biochemistry 2020, 85, 131–153.
- Ponnu, J.; Hoecker, U. Signaling Mechanisms by Arabidopsis Cryptochromes. Front. Plant Sci. 2022, 13, 844714.
- Pooam, M.; El-Esawi, M.A.; Aguida, B.; Ahmad, M. Arabidopsis cryptochrome and Quantum Biology: New insights for plant science and crop improvement. J. Plant Biochem. Biotechnol. 2020, 29, 636–651.
- Brazard, J.; Usman, A.; Lacombat, F.; Ley, C.; Martin, M.M.; Plaza, P.; Mony, L.; Heijde, M.; Zabulon, G.; Bowler, C. Spectro−Temporal Characterization of the Photoactivation Mechanism of Two New Oxidized Cryptochrome/Photolyase Photoreceptors. J. Am. Chem. Soc. 2010, 132, 4935–4945.
- Hense, A.; Herman, E.; Oldemeyer, S.; Kottke, T. Proton transfer to flavin stabilizes the signaling state of the blue light receptor plant cryptochrome. J. Biol. Chem. 2015, 290, 1743–1751.
- Partch, C.L.; Sancar, A. Photochemistry and Photobiology of Cryptochrome Blue-light Photopigments: The Search for a Photocycle. Photochem. Photobiol. 2005, 81, 1291–1304.
- Tan, C.; Guo, L.; Ai, Y.; Li, J.; Wang, L.; Sancar, A.; Luo, Y.; Zhong, D. Direct Determination of Resonance Energy Transfer in Photolyase: Structural Alignment for the Functional State. J. Phys. Chem. A 2014, 118, 10522–10530.
- Vechtomova, Y.; Telegina, T.; Buglak, A.; Kritsky, M. UV Radiation in DNA Damage and Repair Involving DNA-Photolyases and Cryptochromes. Biomedicines 2021, 9, 1564.
- Kiontke, S.; Göbel, T.; Brych, A.; Batschauer, A. DASH-type cryptochromes—Solved and open questions. Biol. Chem. 2020, 401, 1487–1493.
- Lin, C.; Schneps, C.M.; Chandrasekaran, S.; Ganguly, A.; Crane, B.R. Mechanistic insight into light-dependent recognition of Timeless by Drosophila Cryptochrome. Structure 2022, 30, 851–861.e5.
- Palayam, M.; Ganapathy, J.; Guercio, A.M.; Tal, L.; Deck, S.L.; Shabek, N. Structural insights into photoactivation of plant Cryptochrome-2. Commun. Biol. 2021, 4, 28.
- Scheerer, P.; Zhang, F.; Kalms, J.; von Stetten, D.; Krauß, N.; Oberpichler, I.; Lamparter, T. The Class III Cyclobutane Pyrimidine Dimer Photolyase Structure Reveals a New Antenna Chromophore Binding Site and Alternative Photoreduction Pathways. J. Biol. Chem. 2015, 290, 11504–11514.
- Petersen, J.; Rredhi, A.; Szyttenholm, J.; Oldemeyer, S.; Kottke, T.; Mittag, M. The World of Algae Reveals a Broad Variety of Cryptochrome Properties and Functions. Front. Plant Sci. 2021, 12, 766509.
- Chen, S.; Liu, C.; Zhou, C.; Wei, Z.; Li, Y.; Xiong, L.; Yan, L.; Lv, J.; Shen, L.; Xu, L. Identification and characterization of a prokaryotic 6-4 photolyase from Synechococcus elongatus with a deazariboflavin antenna chromophore. Nucleic Acids Res. 2022, 50, 5757–5771.
- Terai, Y.; Sato, R.; Matsumura, R.; Iwai, S.; Yamamoto, J. Enhanced DNA repair by DNA photolyase bearing an artificial light-harvesting chromophore. Nucleic Acids Res. 2020, 48, 10076–10086.
- Geisselbrecht, Y.; Frühwirth, S.; Schroeder, C.; Pierik, A.J.; Klug, G.; Essen, L.-O. CryB from Rhodobacter sphaeroides: A unique class of cryptochromes with new cofactors. EMBO Rep. 2012, 13, 223–229.
- Moon, Y.-J.; Kim, S.I.; Chung, Y.-H. Sensing and responding to UV-A in cyanobacteria. Int. J. Mol. Sci. 2012, 13, 16303–16332.
- Takeda, J.; Nakata, R.; Ueno, H.; Murakami, A.; Iseki, M.; Watanabe, M. Possible involvement of a tetrahydrobiopterin in photoreception for UV-B-induced anthocyanin synthesis in carrot. Photochem. Photobiol. 2014, 90, 1043–1049.
- Moon, Y.-J.; Kim, S.-J.; Park, Y.M.; Chung, Y.-H. Sensing UV/blue: Pterin as a UV-A absorbing chromophore of cryptochrome. Plant Signal. Behav. 2010, 5, 1127–1130.
- Moon, Y.-J.; Lee, E.-M.; Park, Y.M.; Park, Y.S.; Chung, W.-I.; Chung, Y.-H. The role of cyanopterin in UV/blue light signal transduction of cyanobacterium Synechocystis sp. PCC 6803 phototaxis. Plant Cell Physiol. 2010, 51, 969–980.
- Buglak, A.A. Photobiochemistry of Pterin Coenzymes; Research Centre of Biotechnology RAS: Moscow, Russian, 2016.
- Wu, D.; Hu, Q.; Yan, Z.; Chen, W.; Yan, C.; Huang, X.; Zhang, J.; Yang, P.; Deng, H.; Wang, J.; et al. Structural basis of ultraviolet-B perception by UVR8. Nature 2012, 484, 214–219.
- Schallreuter, K.U.; Chavan, B.; Rokos, H.; Hibberts, N.; Panske, A.; Wood, J.M. Decreased phenylalanine uptake and turnover in patients with vitiligo. Mol. Genet. Metab. 2005, 86 (Suppl. 1), S27–S33.
- Niu, C.; Aisa, H.A. Upregulation of Melanogenesis and Tyrosinase Activity: Potential Agents for Vitiligo. Molecules 2017, 22, 1303.
- Telegina, T.A.; Lyudnikova, T.A.; Buglak, A.A.; Vechtomova, Y.L.; Biryukov, M.V.; Demin, V.V.; Kritsky, M.S. Transformation of 6-tetrahydrobiopterin in aqueous solutions under UV-irradiation. J. Photochem. Photobiol. A Chem. 2018, 354, 155–162.
- Telegina, T.A.; Vechtomova, Y.L.; Kritsky, M.S.; Madirov, E.I.; Nizamutdinov, A.S.; Obuhov, Y.N.; Buglak, A.A. Tetrahydrobiopterin Photooxidation: A Key Process in Vitiligo Phototherapy. Appl. Biochem. Microbiol. 2021, 57, 571–578.
- Castaño, C.; Lorente, C.; Martins-Froment, N.; Oliveros, E.; Thomas, A.H. Degradation of α-melanocyte-stimulating hormone photosensitized by pterin. Org. Biomol. Chem. 2014, 12, 3877–3886.
- Davis, M.D.; Kaufman, S.; Milstien, S. Conversion of 6-substituted tetrahydropterins to 7-isomers via phenylalanine hydroxylase-generated intermediates. Proc. Natl. Acad. Sci. USA 1991, 88, 385–389.
- Thöny, B.; Auerbach, G.; Blau, N. Tetrahydrobiopterin biosynthesis, regeneration and functions. Biochem. J. 2000, 347 Pt 1, 1–16.
- Buglak, A.A.; Telegina, T.A.; Vechtomova, Y.L.; Kritsky, M.S. Autoxidation and photooxidation of tetrahydrobiopterin: A theoretical study. Free Radic. Res. 2021, 55, 499–509.
- Wood, J.M.; Schallreuter-Wood, K.U.; Lindsey, N.J.; Callaghan, S.; Gardner, M.L. A specific tetrahydrobiopterin binding domain on tyrosinase controls melanogenesis. Biochem. Biophys. Res. Commun. 1995, 206, 480–485.
- Eichinger, A.; Danecka, M.K.; Möglich, T.; Borsch, J.; Woidy, M.; Büttner, L.; Muntau, A.C.; Gersting, S.W. Secondary BH4 deficiency links protein homeostasis to regulation of phenylalanine metabolism. Hum. Mol. Genet. 2018, 27, 1732–1742.
- Buglak, A.A.; Telegina, T.A.; Lyudnikova, T.A.; Vechtomova, Y.L.; Kritsky, M.S. Photooxidation of tetrahydrobiopterin under UV irradiation: Possible pathways and mechanisms. Photochem. Photobiol. 2014, 90, 1017–1026.
- Schallreuter, K.U.; Wood, J.M.; Körner, C.; Harle, K.M.; Schulz-Douglas, V.; Werner, E.R. 6-Tetrahydrobiopterin functions as a UVB-light switch for de novo melanogenesis. Biochim. Biophys. Acta 1998, 1382, 339–344.
- Jain, A.; Mal, J.; Mehndiratta, V.; Chander, R.; Patra, S.K. Study of oxidative stress in vitiligo. Indian J. Clin. Biochem. 2011, 26, 78–81.
- Wang, Y.; Li, S.; Li, C. Perspectives of New Advances in the Pathogenesis of Vitiligo: From Oxidative Stress to Autoimmunity. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2019, 25, 1017–1023.
- Xuan, Y.; Yang, Y.; Xiang, L.; Zhang, C. The Role of Oxidative Stress in the Pathogenesis of Vitiligo: A Culprit for Melanocyte Death. Oxid. Med. Cell. Longev. 2022, 2022, 8498472.
- Wood, J.M.; Chavan, B.; Hafeez, I.; Schallreuter, K.U. Regulation of tyrosinase by tetrahydropteridines and H2O2. Biochem. Biophys. Res. Commun. 2004, 325, 1412–1417.
- Hasse, S.; Gibbons, N.C.J.; Rokos, H.; Marles, L.K.; Schallreuter, K.U. Perturbed 6-Tetrahydrobiopterin Recycling via Decreased Dihydropteridine Reductase in Vitiligo: More Evidence for H2O2 Stress. J. Investig. Dermatol. 2004, 122, 307–313.
- Rebrin, I.; Bailey, S.W.; Boerth, S.R.; Ardell, M.D.; Ayling, J.E. Catalytic characterization of 4a-hydroxytetrahydropterin dehydratase. Biochemistry 1995, 34, 5801–5810.
- Haavik, J.; Døskeland, A.P.; Flatmark, T. Stereoselective effects in the interactions of pterin cofactors with rat-liver phenylalanine 4-monooxygenase. Eur. J. Biochem. 1986, 160, 1–8.
- Davis, M.D.; Ribeiro, P.; Tipper, J.; Kaufman, S. “7-tetrahydrobiopterin,” a naturally occurring analogue of tetrahydrobiopterin, is a cofactor for and a potential inhibitor of the aromatic amino acid hydroxylases. Proc. Natl. Acad. Sci. USA 1992, 89, 10109–10113.
- Bendall, J.K.; Douglas, G.; McNeill, E.; Channon, K.M.; Crabtree, M.J. Tetrahydrobiopterin in Cardiovascular Health and Disease. Antioxid. Redox Signal. 2013, 20, 3040–3077.
- Schallreuter, K.U.; Wood, J.M.; Ziegler, I.; Lemke, K.R.; Pittelkow, M.R.; Lindsey, N.J.; Gütlich, M. Defective tetrahydrobiopterin and catecholamine biosynthesis in the depigmentation disorder vitiligo. Biochim. Biophys. Acta 1994, 1226, 181–192.
- Estébanez, S.; Lorente, C.; Tosato, M.G.; Miranda, M.A.; Marín, M.L.; Lhiaubet-Vallet, V.; Thomas, A.H. Photochemical formation of a fluorescent thymidine-pterin adduct in DNA. Dye. Pigment. 2019, 160, 624–632.
- Schallreuter, K.U.; Wood, J.M.; Pittelkow, M.R.; Gütlich, M.; Lemke, K.R.; Rödl, W.; Swanson, N.N.; Hitzemann, K.; Ziegler, I. Regulation of melanin biosynthesis in the human epidermis by tetrahydrobiopterin. Science 1994, 263, 1444–1446.
- Dantola, M.L.; Reid, L.O.; Castaño, C.; Lorente, C.; Oliveros, E.; Thomas, A.H. Photosensitization of peptides and proteins by pterin derivatives. Pteridines 2017, 28, 105–114.
- Rokos, H.; Beazley, W.D.; Schallreuter, K.U. Oxidative stress in vitiligo: Photo-oxidation of pterins produces H(2)O(2) and pterin-6-carboxylic acid. Biochem. Biophys. Res. Commun. 2002, 292, 805–811.
- Lorente, C.; Thomas, A.H. Photophysics and photochemistry of pterins in aqueous solution. Acc. Chem. Res. 2006, 39, 395–402.
- Kritsky, M.S.; Telegina, T.A.; Vechtomova, Y.L.; Kolesnikov, M.P.; Lyudnikova, T.A.; Golub, O.A. Excited flavin and pterin coenzyme molecules in evolution. Biochemistry 2010, 75, 1200–1216.
- Heinz, B.; Ried, W.; Dose, K. Thermal Generation of Pteridines and Flavines from Amino Acid Mixtures. Angew. Chemie Int. Ed. 1979, 18, 478–483.
- Heinz, B.; Ried, W. The formation of chromophores through amino acid thermolysis and their possible role as prebiotic photoreceptors. Biosystems 1981, 14, 33–40.
- Schwartz, W. S. W. Fox and K. Dose, Molecular Evolution and the Origin of Life. Z. Allg. Mikrobiol. 1973, 13, 732.
- Kirschning, A. Coenzymes and Their Role in the Evolution of Life. Angew. Chem. Int. Ed. 2021, 60, 6242–6269.
- Kritsky, M.S.; Telegina, T.A.; Lyudnikova, T.A.; Zemskova, Y.L. Coenzymes in Evolution of the RNA World. In Life in the Universe; Seckbach, J., Chela-Flores, J., Owen, T., Raulin, F., Eds.; Springer: Dordrecht, The Netherlands, 2004; pp. 115–118. ISBN 978-94-007-1003-0.
- Hawkins, M.E. Fluorescent pteridine probes for nucleic acid analysis. Methods Enzymol. 2008, 450, 201–231.
- Schmidt, W.; Butler, W.L. Flavin-mediated photoreactions in artificial systems: A possible model for the blue-light photoreceptor pigment in living systems. Photochem. Photobiol. 1976, 24, 71–75.
- Schirrmeister, B.E.; Sanchez-Baracaldo, P.; Wacey, D. Cyanobacterial evolution during the Precambrian. Int. J. Astrobiol. 2016, 15, 187–204.
- Chen, S.-C.; Sun, G.-X.; Yan, Y.; Konstantinidis, K.T.; Zhang, S.-Y.; Deng, Y.; Li, X.-M.; Cui, H.-L.; Musat, F.; Popp, D.; et al. The Great Oxidation Event expanded the genetic repertoire of arsenic metabolism and cycling. Proc. Natl. Acad. Sci. USA 2020, 117, 10414–10421.
- Lee, H.W.; Oh, C.H.; Geyer, A.; Pfleiderer, W.; Park, Y.S. Characterization of a novel unconjugated pteridine glycoside, cyanopterin, in Synechocystis sp. PCC 6803. Biochim. Biophys. Acta 1999, 1410, 61–70.
- Buglak, A.A.; Telegina, T.A. A theoretical study of 5,6,7,8-tetrahydro-6-hydroxymethylpterin: Insight into intrinsic photoreceptor properties of 6-substituted tetrahydropterins. Photochem. Photobiol. Sci. 2019, 18, 516–523.

