 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Juan Zhang | -- | 3150 | 2022-12-12 11:40:58 | | | |
| 2 | Amina Yu | + 27 word(s) | 3177 | 2022-12-13 02:09:27 | | | | |
| 3 | Juan Zhang | Meta information modification | 3177 | 2022-12-13 03:58:36 | | | | |
| 4 | Juan Zhang | Meta information modification | 3177 | 2022-12-13 04:00:03 | | |
Video Upload Options
There are many types of neurodegenerative diseases, and the most common ones are Alzheimer’s disease (AD), Parkinson’s disease (PD), Amyotrophic lateral sclerosis (ALS) and Huntington’s disease (HD). AD is a neuron-centered disease generally characterized by Aβ and tau phosphorylation. PD is generally characterized by progressive deterioration of motor function due to loss of nigrostriatal dopaminergic neurons with muscle rigidity, bradykinesia and resting tremor. ALS is a fatal onset disease characterized by selective loss of upper and lower motor neurons. HD is a predominantly genetic disease, for which there is no drug cure and it is ultimately fatal. Although their underlying mechanisms remain elusive, many studies have revealed that a series of miRNAs are involved in the development of these diseases. MiRNA regulation happens prior to neurological damage, which emphasizes the significance of miRNA alterations in the disease development. Upregulation/downregulation of miRNA expression leads to the alteration of the protein expressed by the corresponding pathogenic gene, which ultimately results in occurrence and development of neurodegenerative diseases.
1. miRNA in Alzheimer’s Disease (AD)
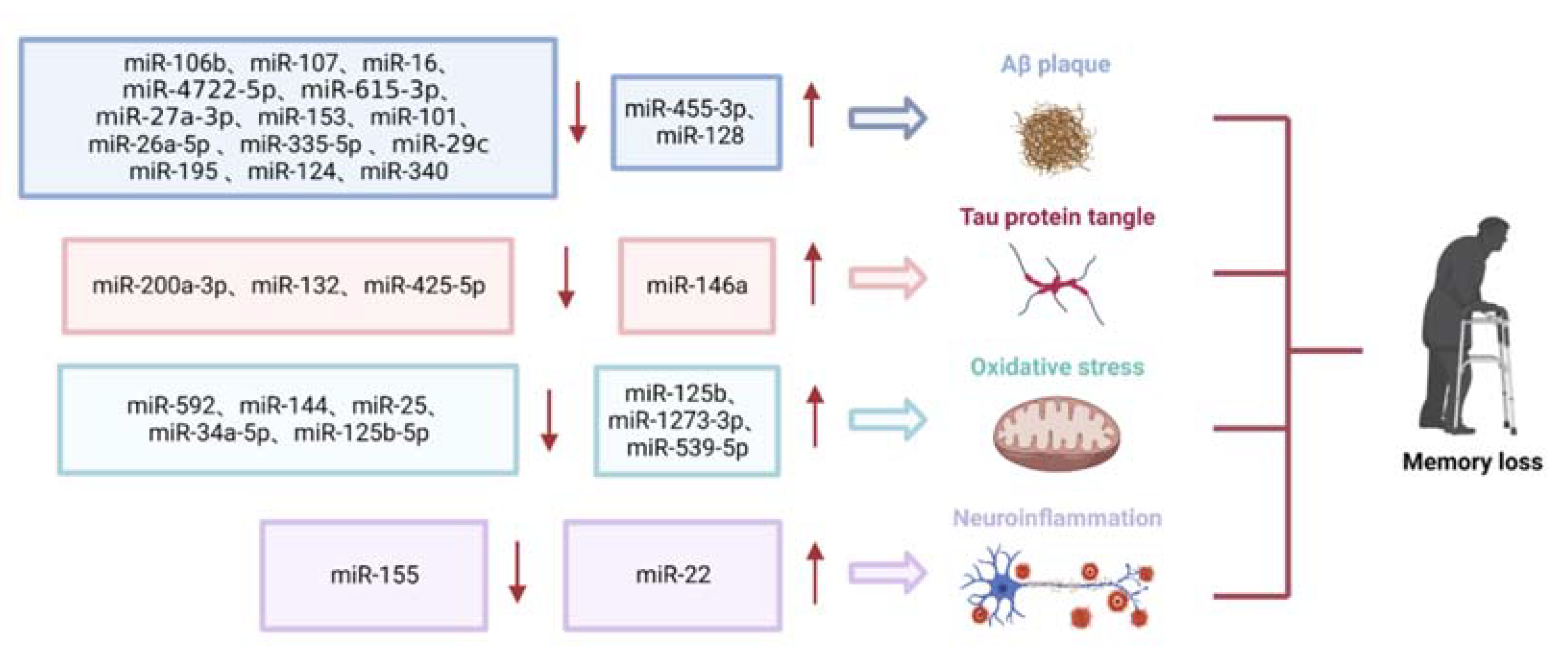
1.1. Role of miRNAs in Aβ Deposition
1.2. Role of miRNAs in Tau Phosphorylation
1.3. Role of miRNAs in Oxidative Stress
1.4. Role of miRNAs in Neuroinflammation
2. miRNA in Parkinson’s Disease (PD)
3. miRNA in Amyotrophic Lateral Sclerosis (ALS)
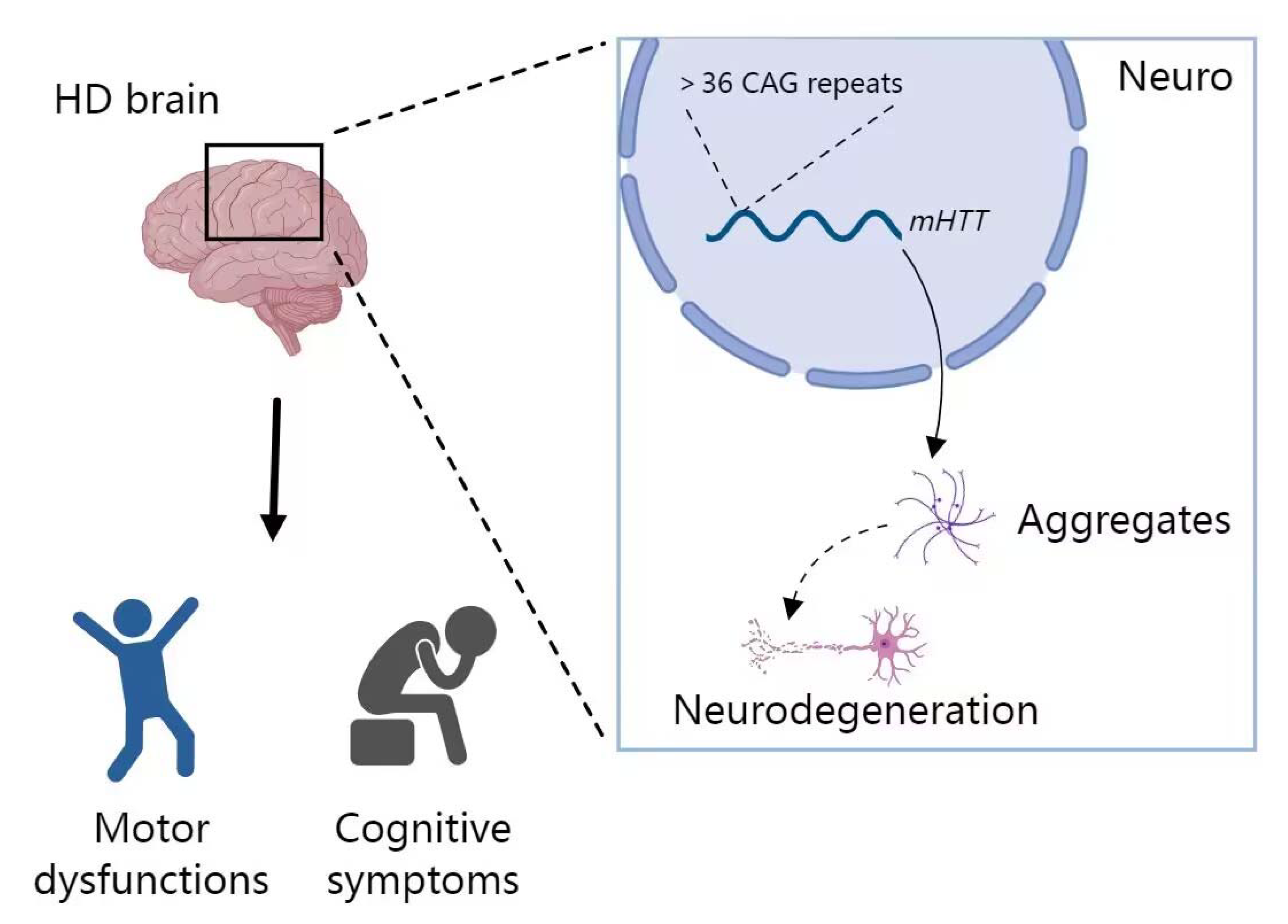
4. miRNA in Huntington’s Disease (HD)
References
- Collaborators, G.D. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 88–106.
- Xu, Y.; Wang, T.; Chen, Z.; Jin, L.; Wu, Z.; Yan, J.; Zhao, X.; Cai, L.; Deng, Y.; Guo, Y.; et al. The point-of-care-testing of nucleic acids by chip, cartridge and paper sensors. Chin. Chem. Lett. 2021, 32, 3675–3686.
- Xiao, C.; Guo, Y.; Zhao, K.; Liu, S.; He, N.; He, Y.; Guo, S.; Chen, Z. Prognostic value of machine learning in patients with acute myocardial infarction. J. Cardiovasc. Dev. Dis. 2022, 9, 56.
- Liu, S.; He, X.; Zhang, T.; Zhao, K.; Xiao, C.; Tong, Z.; Jin, L.; He, N.; Deng, Y.; Li, S.; et al. Highly sensitive smartphone-based detection of listeria monocytogenes using SYTO9. Chin. Chem. Lett. 2022, 33, 1933–1935.
- Chen, Z.; Zhao, K.; He, Z.; Luo, X.; Qin, Z.; Tan, Y.; Zheng, X.; Wu, Z.; Deng, Y.; Chen, H.; et al. Development and evaluation of a thermostatic nucleic acid testing device based on magnesium pyrophosphate precipitation for detecting enterocytozoon hepatopenaei. Chin. Chem. Lett. 2022, 33, 4053–4056.
- Prasad, K.N. Simultaneous activation of Nrf2 and elevation of antioxidant compounds for reducing oxidative stress and chronic inflammation in human Alzheimer’s disease. Mech. Ageing Dev. 2016, 153, 41–47.
- Zhang, M.; Bian, Z. Alzheimer’s disease and microRNA-132: A widespread pathological factor and potential therapeutic target. Front. Neurosci. 2021, 15, 687973.
- Amakiri, N.; Kubosumi, A.; Tran, J.; Reddy, P.H. Amyloid beta and microRNAs in Alzheimer’s disease. Front. Neurosci. 2019, 13, 430.
- Slota, J.A.; Booth, S.A. MicroRNAs in neuroinflammation: Implications in disease pathogenesis, biomarker discovery and therapeutic applications. Noncoding RNA 2019, 5, 35.
- Zhong, Z.; Yuan, K.; Tong, X.; Hu, J.; Song, Z.; Zhang, G.; Fang, X.; Zhang, W. MiR-16 attenuates beta-amyloid-induced neurotoxicity through targeting beta-site amyloid precursor protein-cleaving enzyme 1 in an Alzheimer’s disease cell model. Neuroreport 2018, 29, 1365–1372.
- Goodenough, S.; Schäfer, M.; Behl, C. Estrogen-induced cell signalling in a cellular model of Alzheimer’s disease. J. Steroid Biochem. Mol. Biol. 2003, 84, 301–305.
- Zhang, H.Y.; Zheng, C.Y.; Yan, H.; Wang, Z.F.; Tang, L.L.; Gao, X.; Tang, X.C. Potential therapeutic targets of huperzine a for Alzheimer’s disease and vascular dementia. Chem. Biol. Interact. 2008, 175, 396–402.
- Kim, J.; Yoon, H.; Ramírez, C.M.; Lee, S.-M.; Hoe, H.-S.; Fernández-Hernando, C.; Kim, J. miR-106b impairs cholesterol efflux and increases Aβ levels by repressing ABCA1 expression. Exp. Neurol. 2012, 235, 476–483.
- Liu, Y.; Xu, Y.; Yu, M. MicroRNA-4722-5p and microRNA-615-3p serve as potential biomarkers for Alzheimer’s disease. Exp. Ther. Med. 2022, 23, 241.
- He, L.; Chen, Z.; Wang, J.; Feng, H. Expression relationship and significance of NEAT1 and miR-27a-3p in serum and cerebrospinal fluid of patients with Alzheimer’s disease. BMC Neurol. 2022, 22, 203.
- Ke, S.; Yang, Z.; Yang, F.; Wang, X.; Tan, J.; Liao, B. Long noncoding RNA NEAT1 aggravates Aβ-induced neuronal damage by targeting miR-107 in Alzheimer’s disease. Yonsei Med. J. 2019, 60, 640–650.
- Zhao, M.Y.; Wang, G.Q.; Wang, N.N.; Yu, Q.Y.; Liu, R.L.; Shi, W.Q. The long-non-coding RNA NEAT1 is a novel target for Alzheimer’s disease progression via miR-124/BACE1 axis. Neurol. Res. 2019, 41, 489–497.
- Madadi, S.; Saidijam, M.; Yavari, B.; Soleimani, M. Downregulation of serum miR-106b: A potential biomarker for Alzheimer disease. Arch. Physiol. Biochem. 2022, 128, 875–879.
- Huang, W.; Li, Z.; Zhao, L.; Zhao, W. Simvastatin ameliorate memory deficits and inflammation in clinical and mouse model of Alzheimer’s disease via modulating the expression of miR-106b. Biomed. Pharmacother. 2017, 92, 46–57.
- Wang, W.X.; Rajeev, B.W.; Stromberg, A.J.; Ren, N.; Tang, G.; Huang, Q.; Rigoutsos, I.; Nelson, P.T. The expression of microRNA miR-107 decreases early in Alzheimer’s disease and may accelerate disease progression through regulation of beta-site amyloid precursor protein-cleaving enzyme 1. J. Neurosci. 2008, 28, 1213–1223.
- Nelson, P.T.; Wang, W.X. MiR-107 is reduced in Alzheimer’s disease brain neocortex: Validation study. J. Alzheimers Dis. 2010, 21, 75–79.
- Cheng, J.; Wang, G.; Zhang, N.; Li, F.; Shi, L.; Li, H. Isovitexin modulates autophagy in Alzheimer’s disease via miR-107 signalling. Transl. Neurosci. 2020, 11, 391–401.
- Zong, Y.; Wang, H.; Dong, W.; Quan, X.; Zhu, H.; Xu, Y.; Huang, L.; Ma, C.; Qin, C. miR-29c regulates BACE1 protein expression. Brain Res. 2011, 1395, 108–115.
- Zhu, H.C.; Wang, L.M.; Wang, M.; Song, B.; Tan, S.; Teng, J.F.; Duan, D.X. MicroRNA-195 downregulates Alzheimer’s disease amyloid-beta production by targeting BACE1. Brain Res. Bull. 2012, 88, 596–601.
- Fang, M.; Wang, J.; Zhang, X.; Geng, Y.; Hu, Z.; Rudd, J.A.; Ling, S.; Chen, W.; Han, S. The miR-124 regulates the expression of BACE1/beta-secretase correlated with cell death in Alzheimer’s disease. Toxicol. Lett. 2012, 209, 94–105.
- Liu, W.; Liu, C.; Zhu, J.; Shu, P.; Yin, B.; Gong, Y.; Qiang, B.; Yuan, J.; Peng, X. MicroRNA-16 targets amyloid precursor protein to potentially modulate Alzheimer’s-associated pathogenesis in SAMP8 mice. Neurobiol. Aging 2012, 33, 522–534.
- Liang, C.; Zhu, H.; Xu, Y.; Huang, L.; Ma, C.; Deng, W.; Liu, Y.; Qin, C. MicroRNA-153 negatively regulates the expression of amyloid precursor protein and amyloid precursor-like protein 2. Brain Res. 2012, 1455, 103–113.
- Vilardo, E.; Barbato, C.; Ciotti, M.; Cogoni, C.; Ruberti, F. MicroRNA-101 regulates amyloid precursor protein expression in hippocampal neurons. J. Biol. Chem. 2010, 285, 18344–18351.
- Kumar, S.; Vijayan, M.; Reddy, P.H. MicroRNA-455-3p as a potential peripheral biomarker for Alzheimer’s disease. Hum. Mol. Genet. 2017, 26, 3808–3822.
- Liu, Y.; Wang, L.; Xie, F.; Wang, X.; Hou, Y.; Wang, X.; Liu, J. Overexpression of miR-26a-5p Suppresses tau phosphorylation and abeta accumulation in the Alzheimer’s disease mice by targeting DYRK1A. Curr. Neurovasc. Res. 2020, 17, 241–248.
- Wang, D.; Fei, Z.; Luo, S.; Wang, H. MiR-335-5p inhibits beta-amyloid (abeta) accumulation to attenuate cognitive deficits through targeting c-jun-N-terminal kinase 3 in Alzheimer’s disease. Curr. Neurovasc. Res. 2020, 17, 93–101.
- Tan, X.; Luo, Y.; Pi, D.; Xia, L.; Li, Z.; Tu, Q. MiR-340 reduces the accumulation of amyloid-beta through targeting BACE1 (beta-site amyloid precursor protein cleaving enzyme 1) in Alzheimer’s disease. Curr. Neurovasc. Res. 2020, 17, 86–92.
- Liu, Y.; Zhang, Y.; Liu, P.; Bai, H.; Li, X.; Xiao, J.; Yuan, Q.; Geng, S.; Yin, H.; Zhang, H.; et al. MicroRNA-128 knockout inhibits the development of Alzheimer’s disease by targeting PPARγ in mouse models. Eur. J. Pharmacol. 2019, 843, 134–144.
- Miya Shaik, M.; Tamargo, I.A.; Abubakar, M.B.; Kamal, M.A.; Greig, N.H.; Gan, S.H. The role of microRNAs in Alzheimer’s disease and their therapeutic potentials. Genes 2018, 9, 174.
- Lebouvier, T.; Scales, T.M.; Williamson, R.; Noble, W.; Duyckaerts, C.; Hanger, D.P.; Reynolds, C.H.; Anderton, B.H.; Derkinderen, P. The microtubule-associated protein tau is also phosphorylated on tyrosine. J. Alzheimers Dis. 2009, 18, 1–9.
- Binder, L.I.; Guillozet-Bongaarts, A.L.; Garcia-Sierra, F.; Berry, R.W. Tau, tangles, and Alzheimer’s disease. Biochim. Biophys. Acta 2005, 1739, 216–223.
- Wang, L.; Liu, J.; Wang, Q.; Jiang, H.; Zeng, L.; Li, Z.; Liu, R. MicroRNA-200a-3p mediates neuroprotection in Alzheimer-related deficits and attenuates amyloid-beta overproduction and tau hyperphosphorylation via coregulating BACE1 and PRKACB. Front. Pharmacol. 2019, 10, 806.
- Mancuso, R.; Agostini, S.; Hernis, A.; Zanzottera, M.; Bianchi, A.; Clerici, M. Circulatory miR-223-3p discriminates between Parkinson’s and Alzheimer’s patients. Sci. Rep. 2019, 9, 9393.
- Cha, D.J.; Mengel, D.; Mustapic, M.; Liu, W.; Selkoe, D.J.; Kapogiannis, D.; Galasko, D.; Rissman, R.A.; Bennett, D.A.; Walsh, D.M. miR-212 and miR-132 are downregulated in neurally derived plasma exosomes of Alzheimer’s patients. Front. Neurosci. 2019, 13, 1208.
- Wong, H.K.; Veremeyko, T.; Patel, N.; Lemere, C.A.; Walsh, D.M.; Esau, C.; Vanderburg, C.; Krichevsky, A.M. De-repression of FOXO3a death axis by microRNA-132 and -212 causes neuronal apoptosis in Alzheimer’s disease. Hum. Mol. Genet. 2013, 22, 3077–3092.
- El Fatimy, R.; Li, S.; Chen, Z.; Mushannen, T.; Gongala, S.; Wei, Z.; Balu, D.T.; Rabinovsky, R.; Cantlon, A.; Elkhal, A.; et al. MicroRNA-132 provides neuroprotection for tauopathies via multiple signaling pathways. Acta Neuropathol. 2018, 136, 537–555.
- Deng, Y.; Zhang, J.; Sun, X.; Ma, G.; Luo, G.; Miao, Z.; Song, L. MiR-132 improves the cognitive function of rats with Alzheimer’s disease by inhibiting the MAPK1 signal pathway. Exp. Ther. Med. 2020, 20, 159.
- Yuan, J.; Wu, Y.; Li, L.; Liu, C. MicroRNA-425-5p promotes tau phosphorylation and cell apoptosis in Alzheimer’s disease by targeting heat shock protein B8. J. Neural Transm. 2020, 127, 339–346.
- Mai, H.; Fan, W.; Wang, Y.; Cai, Y.; Li, X.; Chen, F.; Chen, X.; Yang, J.; Tang, P.; Chen, H.; et al. Intranasal administration of miR-146a agomir rescued the pathological process and cognitive impairment in an AD mouse model. Mol. Ther. Nucleic Acids 2019, 18, 681–695.
- Kuchibhotla, K.V.; Goldman, S.T.; Lattarulo, C.R.; Wu, H.-Y.; Hyman, B.T.; Bacskai, B.J. Aβ plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron 2008, 59, 214–225.
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The role of oxidative stress in neurodegenerative diseases. Exp. Neurobiol. 2015, 24, 325–340.
- Chen, X.; Guo, C.; Kong, J. Oxidative stress in neurodegenerative diseases. Neural Regen. Res. 2012, 7, 376–385.
- Konovalova, J.; Gerasymchuk, D.; Parkkinen, I.; Chmielarz, P.; Domanskyi, A. Interplay between microRNAs and oxidative stress in neurodegenerative diseases. Int. J. Mol. Sci. 2019, 20, 6055.
- Jin, Y.; Tu, Q.; Liu, M. MicroRNA-125b regulates Alzheimer’s disease through SphK1 regulation. Mol. Med. Rep. 2018, 18, 2373–2380.
- Wu, G.D.; Li, Z.H.; Li, X.; Zheng, T.; Zhang, D.K. MicroRNA-592 blockade inhibits oxidative stress injury in Alzheimer’s disease astrocytes via the KIAA0319-mediated Keap1/Nrf2/ARE signaling pathway. Exp. Neurol. 2020, 324, 113128.
- Zhou, C.; Zhao, L.; Zheng, J.; Wang, K.; Deng, H.; Liu, P.; Chen, L.; Mu, H. MicroRNA-144 modulates oxidative stress tolerance in SH-SY5Y cells by regulating nuclear factor erythroid 2-related factor 2-glutathione axis. Neurosci. Lett. 2017, 655, 21–27.
- Duan, Q.; Si, E. MicroRNA-25 aggravates Aβ1-42-induced hippocampal neuron injury in Alzheimer’s disease by downregulating KLF2 via the Nrf2 signaling pathway in a mouse model. J. Cell. Biochem. 2019, 120, 15891–15905.
- Kim, S.H.; Choi, K.Y.; Park, Y.; McLean, C.; Park, J.; Lee, J.H.; Lee, K.H.; Kim, B.C.; Huh, Y.H.; Lee, K.H.; et al. Enhanced Expression of microRNA-1273g-3p contributes to Alzheimer’s disease pathogenesis by regulating the expression of mitochondrial Genes. Cells 2021, 10, 2697.
- Jiang, Y.; Zhang, Y.; Su, L. MiR-539-5p decreases amyloid beta-protein production, hyperphosphorylation of tau and memory impairment by regulating PI3K/Akt/GSK-3beta pathways in APP/PS1 double transgenic mice. Neurotox. Res. 2020, 38, 524–535.
- Li, P.; Xu, Y.; Wang, B.; Huang, J.; Li, Q. MiR-34a-5p and miR-125b-5p attenuate abeta-induced neurotoxicity through targeting BACE1. J. Neurol. Sci. 2020, 413, 116793.
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405.
- Xanthos, D.N.; Sandkuhler, J. Neurogenic neuroinflammation: Inflammatory CNS reactions in response to neuronal activity. Nat. Rev. Neurosci. 2014, 15, 43–53.
- Lu, C.E.; Liu, Y.; Sun, B.; Sun, Y.E.; Hou, B.; Zhang, Y.; Ma, Z.; Gu, X. Intrathecal injection of JWH-015 attenuates bone cancer pain via time-dependent modification of pro-inflammatory cytokines expression and astrocytes activity in spinal cord. Inflammation 2015, 38, 1880–1890.
- Craft, J.M.; Watterson, D.M.; Van Eldik, L.J. Human amyloid beta-induced neuroinflammation is an early event in neurodegeneration. Glia 2006, 53, 484–490.
- Pizza, V.; Agresta, A.; D’Acunto, C.W.; Festa, M.; Capasso, A. Neuroinflamm-aging and neurodegenerative diseases: An overview. CNS Neurol. Disord. Drug Targets 2011, 10, 621–634.
- Varnum, M.M.; Ikezu, T. The classification of microglial activation phenotypes on neurodegeneration and regeneration in Alzheimer’s disease brain. Arch. Immunol. Ther. Exp. 2012, 60, 251–266.
- Cagnin, A.; Brooks, D.J.; Kennedy, A.M.; Gunn, R.N.; Myers, R.; Turkheimer, F.E.; Jones, T.; Banati, R.B. In-vivo measurement of activated microglia in dementia. Lancet 2001, 358, 461–467.
- Fillit, H.; Ding, W.H.; Buee, L.; Kalman, J.; Altstiel, L.; Lawlor, B.; Wolf-Klein, G. Elevated circulating tumor necrosis factor levels in Alzheimer’s disease. Neurosci. Lett. 1991, 129, 318–320.
- Tan, M.S.; Yu, J.T.; Jiang, T.; Zhu, X.C.; Tan, L. The NLRP3 inflammasome in Alzheimer’s disease. Mol. Neurobiol. 2013, 48, 875–882.
- Tarassishin, L.; Loudig, O.; Bauman, A.; Shafit-Zagardo, B.; Suh, H.S.; Lee, S.C. Interferon regulatory factor 3 inhibits astrocyte inflammatory gene expression through suppression of the proinflammatory miR-155 and miR-155*. Glia 2011, 59, 1911–1922.
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158.
- Voet, S.; Srinivasan, S.; Lamkanfi, M.; van Loo, G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. 2019, 11, e10248.
- Han, C.; Guo, L.; Yang, Y.; Guan, Q.; Shen, H.; Sheng, Y.; Jiao, Q. Mechanism of microRNA-22 in regulating neuroinflammation in Alzheimer’s disease. Brain Behav. 2020, 10, e01627.
- Lesage, S.; Brice, A. Parkinson’s disease: From monogenic forms to genetic susceptibility factors. Hum. Mol. Genet. 2009, 18, R48–R59.
- Leitão, A.D.G.; Rudolffi-Soto, P.; Chappard, A.; Bhumkar, A.; Lau, D.; Hunter, D.J.B.; Gambin, Y.; Sierecki, E. Selectivity of Lewy body protein interactions along the aggregation pathway of α-synuclein. Commun. Biol. 2021, 4, 1124.
- Khezri, M.R.; Yousefi, K.; Zolbanin, N.M.; Ghasemnejad-Berenji, M. MicroRNAs in the pathophysiology of Alzheimer’s disease and Parkinson’s disease: An overview. Mol. Neurobiol. 2022, 59, 1589–1603.
- Briggs, C.E.; Wang, Y.; Kong, B.; Woo, T.U.; Iyer, L.K.; Sonntag, K.C. Midbrain dopamine neurons in Parkinson’s disease exhibit a dysregulated miRNA and target-gene network. Brain Res. 2015, 1618, 111–121.
- Burré, J. The synaptic function of α-synuclein. J. Park. Dis. 2015, 5, 699–713.
- Xu, L.; Pu, J. Alpha-synuclein in Parkinson’s Disease: From pathogenetic dysfunction to potential clinical application. Park. Dis. 2016, 2016, 1720621.
- Lashuel, H.A.; Petre, B.M.; Wall, J.; Simon, M.; Nowak, R.J.; Walz, T.; Lansbury, P.T., Jr. Alpha-synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. J. Mol. Biol. 2002, 322, 1089–1102.
- Wang, Z.H.; Zhang, J.L.; Duan, Y.L.; Zhang, Q.S.; Li, G.F.; Zheng, D.L. MicroRNA-214 participates in the neuroprotective effect of Resveratrol via inhibiting α-synuclein expression in MPTP-induced Parkinson’s disease mouse. Biomed. Pharmacother. 2015, 74, 252–256.
- Sahay, S.; Ghosh, D.; Singh, P.K.; Maji, S.K. Alteration of structure and aggregation of α-synuclein by familial Parkinson’s disease associated mutations. Curr. Protein Pept. Sci. 2017, 18, 656–676.
- Yang, Y.; Li, Y.; Yang, H.; Guo, J.; Li, N. Circulating microRNAs and long non-coding RNAs as potential diagnostic biomarkers for Parkinson’s disease. Front. Mol. Neurosci. 2021, 14, 631553.
- Nies, Y.H.; Mohamad Najib, N.H.; Lim, W.L.; Kamaruzzaman, M.A.; Yahaya, M.F.; Teoh, S.L. MicroRNA dysregulation in Parkinson’s disease: A Narrative Review. Front. Neurosci. 2021, 15, 660379.
- Evans, B.; Furlong, H.A.T.; de Lencastre, A. Parkinson’s disease and microRNAs—Lessons from model organisms and human studies. Exp. Gerontol. 2021, 155, 111585.
- Doxakis, E. Post-transcriptional regulation of alpha-synuclein expression by mir-7 and mir-153. J. Biol. Chem. 2010, 285, 12726–12734.
- Wu, Y.Y.; Kuo, H.C. Functional roles and networks of non-coding RNAs in the pathogenesis of neurodegenerative diseases. J. Biomed. Sci. 2020, 27, 49.
- McMillan, K.J.; Murray, T.K.; Bengoa-Vergniory, N.; Cordero-Llana, O.; Cooper, J.; Buckley, A.; Wade-Martins, R.; Uney, J.B.; O’Neill, M.J.; Wong, L.F.; et al. Loss of microRNA-7 regulation leads to α-synuclein accumulation and dopaminergic neuronal loss in vivo. Mol. Ther. 2017, 25, 2404–2414.
- Consales, C.; Cirotti, C.; Filomeni, G.; Panatta, M.; Butera, A.; Merla, C.; Lopresto, V.; Pinto, R.; Marino, C.; Benassi, B. Fifty-hertz magnetic field affects the epigenetic modulation of the miR-34b/c in neuronal cells. Mol. Neurobiol. 2018, 55, 5698–5714.
- Tarale, P.; Daiwile, A.P.; Sivanesan, S.; Stöger, R.; Bafana, A.; Naoghare, P.K.; Parmar, D.; Chakrabarti, T.; Krishnamurthi, K. Manganese exposure: Linking down-regulation of miRNA-7 and miRNA-433 with α-synuclein overexpression and risk of idiopathic Parkinson’s disease. Toxicol. In Vitro 2018, 46, 94–101.
- Kabaria, S.; Choi, D.C.; Chaudhuri, A.D.; Mouradian, M.M.; Junn, E. Inhibition of miR-34b and miR-34c enhances α-synuclein expression in Parkinson’s disease. FEBS Lett. 2015, 589, 319–325.
- Verma, M.; Steer, E.K.; Chu, C.T. ERKed by LRRK2: A cell biological perspective on hereditary and sporadic Parkinson’s disease. Biochim. Biophys. Acta. 2014, 1842, 1273–1281.
- Esteves, A.R.; Swerdlow, R.H.; Cardoso, S.M. LRRK2, a puzzling protein: Insights into Parkinson’s disease pathogenesis. Exp. Neurol. 2014, 261, 206–216.
- Gehrke, S.; Imai, Y.; Sokol, N.; Lu, B. Pathogenic LRRK2 negatively regulates microRNA-mediated translational repression. Nature 2010, 466, 637–641.
- Cho, H.J.; Liu, G.; Jin, S.M.; Parisiadou, L.; Xie, C.; Yu, J.; Sun, L.; Ma, B.; Ding, J.; Vancraenenbroeck, R.; et al. MicroRNA-205 regulates the expression of Parkinson’s disease-related leucine-rich repeat kinase 2 protein. Hum. Mol. Genet. 2013, 22, 608–620.
- Wu, Q.; Xi, D.Z.; Wang, Y.H. MicroRNA-599 regulates the development of Parkinson’s disease through mediating LRRK2 expression. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 724–731.
- Ricci, C.; Marzocchi, C.; Battistini, S. MicroRNAs as biomarkers in Amyotrophic lateral sclerosis. Cells 2018, 7, 219.
- Henriques, A.; Pitzer, C.; Schneider, A. Neurotrophic growth factors for the treatment of amyotrophic lateral sclerosis: Where do we stand? Front. Neurosci. 2010, 4, 32.
- Bucchia, M.; Ramirez, A.; Parente, V.; Simone, C.; Nizzardo, M.; Magri, F.; Dametti, S.; Corti, S. Therapeutic development in Amyotrophic lateral sclerosis. Clin. Ther. 2015, 37, 668–680.
- Cloutier, F.; Marrero, A.; O’Connell, C.; Morin, P., Jr. MicroRNAs as potential circulating biomarkers for Amyotrophic lateral sclerosis. J. Mol. Neurosci. 2015, 56, 102–112.
- Dardiotis, E.; Aloizou, A.M.; Siokas, V.; Patrinos, G.P.; Deretzi, G.; Mitsias, P.; Aschner, M.; Tsatsakis, A. The role of microRNAs in patients with Amyotrophic lateral sclerosis. J. Mol. Neurosci. 2018, 66, 617–628.
- Kovanda, A.; Leonardis, L.; Zidar, J.; Koritnik, B.; Dolenc-Groselj, L.; Ristic Kovacic, S.; Curk, T.; Rogelj, B. Differential expression of microRNAs and other small RNAs in muscle tissue of patients with ALS and healthy age-matched controls. Sci. Rep. 2018, 8, 5609.
- Sumitha, R.; Sidhu, R.J.; Sathyaprabha, T.N.; Nalini, A.; Raju, T.R.; Alladi, P.A. Differential expression of microRNA-206 in the gastrocnemius and biceps brachii in response to CSF from sporadic amyotrophic lateral sclerosis patients. J. Neurol. Sci. 2014, 345, 254–256.
- Rastegar-Moghaddam, S.H.; Ebrahimzadeh-Bideskan, A.; Shahba, S.; Malvandi, A.M.; Mohammadipour, A. MicroRNA-22: A novel and potent biological therapeutics in neurological disorders. Mol. Neurobiol. 2022, 59, 2694–2701.
- Williams, A.H.; Valdez, G.; Moresi, V.; Qi, X.; McAnally, J.; Elliott, J.L.; Bassel-Duby, R.; Sanes, J.R.; Olson, E.N. MicroRNA-206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science 2009, 326, 1549–1554.
- Koval, E.D.; Shaner, C.; Zhang, P.; du Maine, X.; Fischer, K.; Tay, J.; Chau, B.N.; Wu, G.F.; Miller, T.M. Method for widespread microRNA-155 inhibition prolongs survival in ALS-model mice. Hum. Mol. Genet. 2013, 22, 4127–4135.
- Hawley, Z.C.E.; Campos-Melo, D.; Strong, M.J. MiR-105 and miR-9 regulate the mRNA stability of neuronal intermediate filaments. Implications for the pathogenesis of Amyotrophic lateral sclerosis (ALS). Brain Res. 2019, 1706, 93–100.
- Kocerha, J.; Xu, Y.; Prucha, M.S.; Zhao, D.; Chan, A.W. MicroRNA-128a dysregulation in transgenic Huntington’s disease monkeys. Mol. Brain. 2014, 7, 46.
- Chu, E.M.; O’Neill, M.; Purkayastha, D.D.; Knight, C. Huntington’s disease: A forensic risk factor in women. J. Clin. Mov. Disord. 2019, 6, 3.
- Li, H.L.; Li, X.Y.; Dong, Y.; Zhang, Y.B.; Cheng, H.R.; Gan, S.R.; Liu, Z.J.; Ni, W.; Burgunder, J.M.; Yang, X.W.; et al. Clinical and genetic profiles in chinese patients with Huntington’s Disease: A ten-year multicenter study in china. Aging Dis. 2019, 10, 1003–1011.
- Li, Q.; Li, G.; Wu, D.; Lu, H.; Hou, Z.; Ross, C.A.; Yang, Y.; Zhang, J.; Duan, W. Resting-state functional MRI reveals altered brain connectivity and its correlation with motor dysfunction in a mouse model of Huntington’s disease. Sci Rep. 2017, 7, 16742.
- Lim, S.A.O.; Surmeier, D.J. Enhanced GABAergic inhibition of cholinergic interneurons in the zQ175(+/-) mouse model of Huntington’s Disease. Front Syst. Neurosci. 2020, 14, 626412.
- Shin, H.; Kim, M.H.; Lee, S.J.; Lee, K.H.; Kim, M.J.; Kim, J.S.; Cho, J.W. Decreased metabolism in the cerebral cortex in early-stage Huntington’s disease: A possible biomarker of disease progression? J. Clin. Neurol. 2013, 9, 21–25.
- Gil, J.M.; Rego, A.C. Mechanisms of neurodegeneration in Huntington’s disease. Eur. J. Neurosci. 2008, 27, 2803–2820.
- Teixeira, A.L.; de Souza, L.C.; Rocha, N.P.; Furr-Stimming, E.; Lauterbach, E.C. Revisiting the neuropsychiatry of Huntington’s disease. Dement Neuropsychol. 2016, 10, 261–266.
- Solberg, O.K.; Filkukova, P.; Frich, J.C.; Feragen, K.J.B. Age at death and causes of death in patients with Huntington disease in norway in 1986-2015. J. Huntingtons. Dis. 2018, 7, 77–86.
- Kachian, Z.R.; Cohen-Zimerman, S.; Bega, D.; Gordon, B.; Grafman, J. Suicidal ideation and behavior in Huntington’s disease: Systematic review and recommendations. J. Affect. Disord. 2019, 250, 319–329.
- Hoss, A.G.; Lagomarsino, V.N.; Frank, S.; Hadzi, T.C.; Myers, R.H.; Latourelle, J.C. Study of plasma-derived miRNAs mimic differences in Huntington’s disease brain. Mov. Disord. 2015, 30, 1961–1964.
- Reed, E.R.; Latourelle, J.C.; Bockholt, J.H.; Bregu, J.; Smock, J.; Paulsen, J.S.; Myers, R.H.; PREDICT-HD CSF Ancillary Study Investigators. MicroRNAs in CSF as prodromal biomarkers for Huntington disease in the PREDICT-HD study. Neurology 2018, 90, e264–e272.
- Shah, R.; Lee, S.C.; Strasser, R.B.; Grossman, C. An Australian neuro-palliative perspective on Huntington’s disease: A case report. BMC Palliat Care 2021, 20, 53.
- Carbo, M.; Brandi, V.; Pascarella, G.; Staid, D.S.; Colotti, G.; Polticelli, F.; Ilari, A.; Morea, V. Bioinformatics analysis of ras homologue enriched in the striatum, a potential target for Huntington’s disease therapy. Int. J. Mol. Med. 2019, 44, 2223–2233.
- Hu, J.; Liu, J.; Yu, D.; Aiba, Y.; Lee, S.; Pendergraff, H.; Boubaker, J.; Artates, J.W.; Lagier-Tourenne, C.; Lima, W.F.; et al. Exploring the effect of sequence length and composition on allele-selective inhibition of human huntingtin expression by single-stranded silencing RNAs. Nucleic Acid. Ther. 2014, 24, 199–209.
- Hoss, A.G.; Kartha, V.K.; Dong, X.; Latourelle, J.C.; Dumitriu, A.; Hadzi, T.C.; Macdonald, M.E.; Gusella, J.F.; Akbarian, S.; Chen, J.F.; et al. MicroRNAs located in the Hox gene clusters are implicated in Huntington’s disease pathogenesis. PLoS Genet. 2014, 10, e1004188.
- Martí, E.; Pantano, L.; Bañez-Coronel, M.; Llorens, F.; Miñones-Moyano, E.; Porta, S.; Sumoy, L.; Ferrer, I.; Estivill, X. A myriad of miRNA variants in control and Huntington’s disease brain regions detected by massively parallel sequencing. Nucleic Acids Res. 2010, 38, 7219–7235.
- Chang, K.H.; Wu, Y.R.; Chen, C.M. Down-regulation of miR-9* in the peripheral leukocytes of Huntington’s disease patients. Orphanet J. Rare Dis. 2017, 12, 185.
- Kumar, S.; Vijayan, M.; Bhatti, J.S.; Reddy, P.H. MicroRNAs as peripheral biomarkers in aging and age-related diseases. Prog. Mol. Biol. Transl. Sci. 2017, 146, 47–94.
- Viswambharan, V.; Thanseem, I.; Vasu, M.M.; Poovathinal, S.A.; Anitha, A. miRNAs as biomarkers of neurodegenerative disorders. Biomark. Med. 2017, 11, 151–167.
- Reynolds, R.H.; Petersen, M.H.; Willert, C.W.; Heinrich, M.; Nymann, N.; Dall, M.; Treebak, J.T.; Bjorkqvist, M.; Silahtaroglu, A.; Hasholt, L.; et al. Perturbations in the p53/miR-34a/SIRT1 pathway in the R6/2 Huntington’s disease model. Mol. Cell. Neurosci. 2018, 88, 118–129.
- Soldati, C.; Bithell, A.; Johnston, C.; Wong, K.Y.; Stanton, L.W.; Buckley, N.J. Dysregulation of REST-regulated coding and non-coding RNAs in a cellular model of Huntington’s disease. J. Neurochem. 2013, 124, 418–430.
- Johnson, R.; Zuccato, C.; Belyaev, N.D.; Guest, D.J.; Cattaneo, E.; Buckley, N.J. A microRNA-based gene dysregulation pathway in Huntington’s disease. Neurobiol. Dis. 2008, 29, 438–445.
- Packer, A.N.; Xing, Y.; Harper, S.Q.; Jones, L.; Davidson, B.L. The bifunctional microRNA miR-9/miR-9* regulates REST and CoREST and is downregulated in Huntington’s disease. J. Neurosci. 2008, 28, 14341–14346.
- Johnson, R.; Buckley, N.J. Gene dysregulation in Huntington’s disease: REST, microRNAs and beyond. Neuromol. Med. 2009, 11, 183–199.



