Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Andre H B Dourado | -- | 2893 | 2022-12-07 17:32:57 | | | |
| 2 | Dean Liu | -3 word(s) | 2890 | 2022-12-09 09:43:55 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Dourado, A.H.B. Electric Double Layer Structure. Encyclopedia. Available online: https://encyclopedia.pub/entry/38369 (accessed on 23 July 2026).
Dourado AHB. Electric Double Layer Structure. Encyclopedia. Available at: https://encyclopedia.pub/entry/38369. Accessed July 23, 2026.
Dourado, André H. B.. "Electric Double Layer Structure" Encyclopedia, https://encyclopedia.pub/entry/38369 (accessed July 23, 2026).
Dourado, A.H.B. (2022, December 08). Electric Double Layer Structure. In Encyclopedia. https://encyclopedia.pub/entry/38369
Dourado, André H. B.. "Electric Double Layer Structure." Encyclopedia. Web. 08 December, 2022.
Copy Citation
The electric double layer (EDL) is the most important electrochemical and heterogeneous catalysis region. Because of it, its modeling and investigation are something that can be found in the literature for a long time. However, it is still something in debate, since nowadays a series of new techniques are available for the investigation of this interfacial area at the molecular level by experiments and simulations.
electric double layer
double layer structure
electrocatalysis
1. Introduction
The electric double layer (EDL) is the most important part of any electrochemical system because it is the region in space where the electrochemical reaction takes place. It was always understood that the composition and conformation of this interfacial region are important to the electrochemical reaction [1][2][3][4][5][6]. The composition, thickness, conformation, and other parameters are already expected to be dependent on the electrode material, the electrolyte chemistry, and the interaction between these two phases. Because of it, the development of electrochemical theory always considered these factors for modeling the electrochemical reactions, but just recently (around the last 30 years), the electrochemical information could merge with molecular-level information, and the EDL structure could be investigated [7][8][9][10][11][12][13][14][15][16][17][18].
Most of the works in the literature investigated the EDL response to the applied potential of polarized electrodes without the presence of faradaic reaction, which allowed the investigation of how the capacitive current is dependent on EDL, [7][10][11] a matter of importance for double layer capacitor devices, [19][20][21][22] in which also some quantum effects can also be present for enhancing the capacitance: [23][24][25] electric double layer transistors, [26] as well as for electrocatalysts, e.g., However, some authors also presented information under the presence of faradaic processes and suggested that the solvent orientation, as well as supporting electrolyte ions adsorption are of great importance to the stabilization of key intermediates [16][17][27][28][29][30][31].
2. EDL Structure
For the reader that is not familiarized with the classic EDL models, such as the Helmholtz or Gouy-Chapman, or Stern models, please go to the review that originates this entry, where full details are given. In there, the relation between EDL and electrochemical measurements and kinetics is also presented. The present text will be focused on the molecular interpretation of EDL structure, which can contribute to adsorbed species stability through intermolecular interactions [28][29][32][33]. To better comprehend it, spectroscopic techniques and theoretical calculations need to be used for these insights. The present work will focus on experimental approaches for the EDL structure; however, some theoretical comparisons will be done when significant. For the reader interested in theoretical/molecular simulations, the works by Wu [23], Chu [34], and Rossmeisl [35][36][37][38][39][40] are highly recommended.
To investigate electrochemical interfaces by experimental approaches, coupling the electrochemical measurement with a spectroscopic technique is needed. Along the most used spectroelectrochemical approaches [35][41][42][43][44][45][46][47][48], the vibrational ones are the most used for EDL investigation due to their traditional application for intermediate/product identification and mechanistic proposals [17][28][29][43][44][45][46][49][50][51][52][53][54]. Because of that, vibrational spectroelectrochemistry is the main focus of this section. The operational principles, selection rules, and limitations of infrared and/or Raman surface/electrochemical spectroscopy are not discussed in here.
Before considering electrochemical reactions, it is better to consider how the EDL can respond to small potential perturbations. In this sense, works using surface-enhanced infrared spectroscopy (SEIRAS) [7][10][43][55][56], surface-enhanced Raman spectroscopy (SERS) [57], and shell-isolated nanoparticle-enhanced Raman spectroscopy (SHINERS) [11] can be found. For these studies, single-crystal electrodes were the most used ones, since they should make the electrode material contribution constant for the whole analysis [12][31][35].
Independent of the supporting electrolyte used, water bands are always present. Because of it, the water-related bands are the target ones for EDL investigations. The banding band (δH2O, around 1600 cm−1) is a qualitative guide for surface wetting [17][54][58]. For most cases, this band is normally not centered at the expected region for free water molecules (1645 cm−1), and when it is redshifted to below 1600 cm−1, the shift is explained by the fact that the inner Helmholtz plane (IHP) water is stabilized by the interaction with the electrode surface [7][10]. In acidic media, it is necessary to consider that the hydronium species can also co-adsorb water. This interaction between hydronium and water generates a double degenerated asymmetric banding centered around 1700 cm−1 [7][8][9][10][11][27][29][55]. The increment in the hydronium-related band can be also related to the surface excess charge; in other words, if the electrode is more negatively charged, the IHP and outer Helmholtz plane (OHP) hydronium concentrations increase, and the 1700 cm−1 intensity increases. In this way, the behavior of this band is normally related to a spectroscopic determination of the potential of zero charge (PZC) [7][8][9][10][11].
For neutral conditions, the shifts in the δH2O can be related to different conformations, such as the weakly adsorbed water molecule, with the interaction by the H atoms (H-down, Figure 1A) expected to be around 1615 cm−1 [7][8][9][10], the bi-layered conformation related to the “ice-like” structure (Figure 1B) around 1640 cm−1 [7][10], and the stronger interaction by the oxygen electrons lone-pairs and the surface at 1650 cm−1 (H-up, Figure 1C) [10].

Figure 1. Schematics of water adsorbed structures on electrode surfaces (red rectangles). Red spheres represent oxygen atoms, black hydrogen, and yellow sulfur: (A) H-down conformation observed at potentials more negative than the PZC [7][8][9]; (B) “ice-like” bilayer structure observed around PZC [7][8][9][10]. (C) H-up conformation observed at potentials more positive than the PZC [7][8][9]; (D) solvated sulfate anions suggested for positive potentials [10]; (E) water dimer; (F) adsorbed OH− [59]; (G) flat adsorbed water at potentials around PZC [7][10].
Another band observed for water molecules is the O-H stretching (νOH). This is very intense, and the broadband is localized over 3000–3200 cm−1. In most spectroelectrochemical investigations, this band is just cited as a “water-related band” and no further interest is given to it [49][51][60][61][62]. However, it is already known that this is not a fully symmetric band; it presents several shoulders, which sometimes evolve into proper bands [7][10]. Applying deconvolution methods, the contribution of smaller bands can also be investigated, such as the one centered around 2900 cm−1, and attributed to the stretching of an O-H moiety in which the H atom is also interacting with the electrode surface in the IHP (H-down, Figure 1A) [7], 3250 cm−1, related to a Fermi resonance of νOH with a δH2O overtone [7]. At 3400 cm−1, the band is suggested to be related to the solvation shell of anions, mainly SO42− (Figure 1D) [8][9]. At 3470 cm−1, the νOH is related to an almost flat-water molecule conformation (Figure 1C); however, it is a little tilted, a condition needed for the observation of this band due to the surface selection rules, and the shift is interpreted to be related to an interaction between the O lone-pairs and the surface [8][9]. At higher wavenumbers, such as 3640 and 3735 cm−1, water dimers are expected (Figure 1E) [7], while for alkaline media, the νOH for hydroxyl ions is likely to be at 3750 cm−1 (Figure 1F) [59].
The bi-layered “ice-like” structure (Figure 1B), also presents a characteristic νOH, which can be observed around 3200 cm−1, according to Garcia-Araez et al. [9] or around 3612 cm−1, according to Ataka et al. [7] In this conformation, just one water layer is related to the IHP, while the second is already considered as part of the OHP [35]. The naming of this structure as “ice-like” is supported by the fact that its adsorption strength is close to the bulk ice binding energy. Additionally, at this conformation, no chemical interaction between the IHP water (first layer) and the electrode is observed, purely physical adsorption [35].
At the PZC, the EDL water is reported to be closer to bulk conformation [11], presenting no preferential interaction, just a flat relation having one lone-pair interacting with the electrode (Figure 1G) [7][56] or even the “ice-like” conformation (Figure 1B) [10]. Ataka et al. [7] proposed that at potentials more negative than the PZC, δH2O should be around 1612 cm−1 and is related to a weakly H-bonded molecule and to a conformation in which the H-down conformation (Figure 1A) is not fully perpendicular (at a 60o tilt); so, there is an interaction between the metallic surface and an oxygen lone-pair [7][8][9]. Wandlowski et al. [10], at the same conditions, also suggested that a second layer is related to the H-bond between the IHP tilted water, and an OHP water is present by also looking at the νOH bands. At the PZC (≈0.58 VRHE, Au electrode in 0.5 mol L−1 HClO4 electrolyte [7]), no preference was observed, and the spectrum at this potential was used as a background [7][8][9][10][11].
At potentials shortly more positive than the PZC, Ataka et al. [7] and Wandlowski et al. [10] suggested the “ice-like” conformation (Figure 1B). However, the range in which each group suggested such a structure did not match, and later, Garcia-Araez et al. [9] highlighted that the divergence between both excellent works could be due to the proposed PZC region. At even more positive potentials, the electrode is expected to be fully H-up (Figure 1C), and some anions are expected to co-adsorb as well (Figure 1D); so, water bands in the solvation shell are also expected [7][8][9][10][28][29][56].
At potentials more negative than the PZC, one of the most interesting works is using SHINERS [11]. There, the authors observed that two possible H-down conformations can be observed. The first is related to a row of water molecules in which one H is interacting with the electrode (Figure 2A), while the other is involved in an H-bond with the water molecule directly in front; the second conformation is also a bi-layered one (Figure 2B) in which the IHP water has both H-atoms interacting with the electrode, while the OHP molecules have one in an extended interaction with the electrode and the second is an H-bond with the IHP molecules.
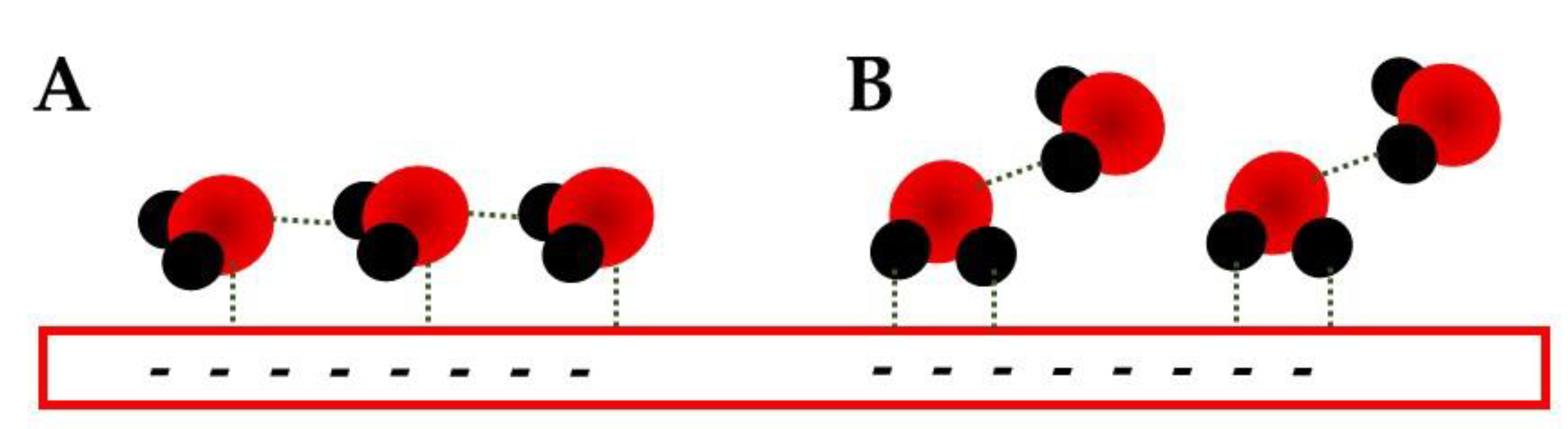
Figure 2. Representation of the two H-down conformation described by Li et al. [11] at potentials more negative than PZC; (A) is expected for shortly more negative than the PZC and (B) for potentials much more negative.
One of the biggest issues of investigating EDL by vibrational spectroscopy is that it is difficult to interpret the relation between band position and potential [11][27][55]. The literature highlights that it can be a Stark shifting, related to how strongly adsorbed the water conformation of one or the other IHP is due to the changing in the conformation, which normally has bands closely centered. For this reason, the investigation of these bands needs to be taken with care and always considering both moieties, δH2O, and νOH.
In electrolytic solutions, water is not the only chemical species present, there are also ionic ones. In acidic media, the hydronium ions (δH3O+ around 1700 cm−1) are the cation, and the anions are, frequently, sulfate [9][10][17][18][29][53][55], perchlorate [7][8][9][17][28][29], and sometimes other acids, such as nitric [18] and halogenates [57], which are also present. The anion participation in the EDL structure is very important, especially for oxidative reactions since they normally occur at potentials more positive than the PZC. These anions are attracted/repelled to/from the EDL by the surface charging process and may assume different conformations [17][18].
The kosmotropic anions, such as sulfate, are known to strongly interact with the surface [63][64][65], and this may shift the PZC, as discussed previously. The PZC shift is normally reflected in a positive shift in the oxidation potential, making the oxidation process less favorable [28][29]. The strong interaction may also block some catalytic sites, and because of it, the electrocatalytic overall current may be smaller. The bands related to sulfate are around 1100 cm−1 [7][9][10][17][18][54], and this band can be deconvoluted in two C2v vibration modes related to adsorption in which two oxygen atoms are interacting with the surface (Figure 3A) [17][18]. This conformation results in two observed bands, one above 1100 cm−1, which is the stronger one, and another below 1000 cm−1. The first is related to the asymmetric stretching and the other to the symmetric one [18]. Another possible conformation for the sulfate adsorption is related to an inverted tetrahedron, in which just one O atom is interacting with the surface in a C3v mode (Figure 3B). The symmetric stretching is expected to be between 750 and 900 cm−1 and the asymmetric between 1250–1350 cm−1 [18]. All these bands are expected to be observed as a broad and an asymmetric band, so once again deconvolution methods are recommended [17].
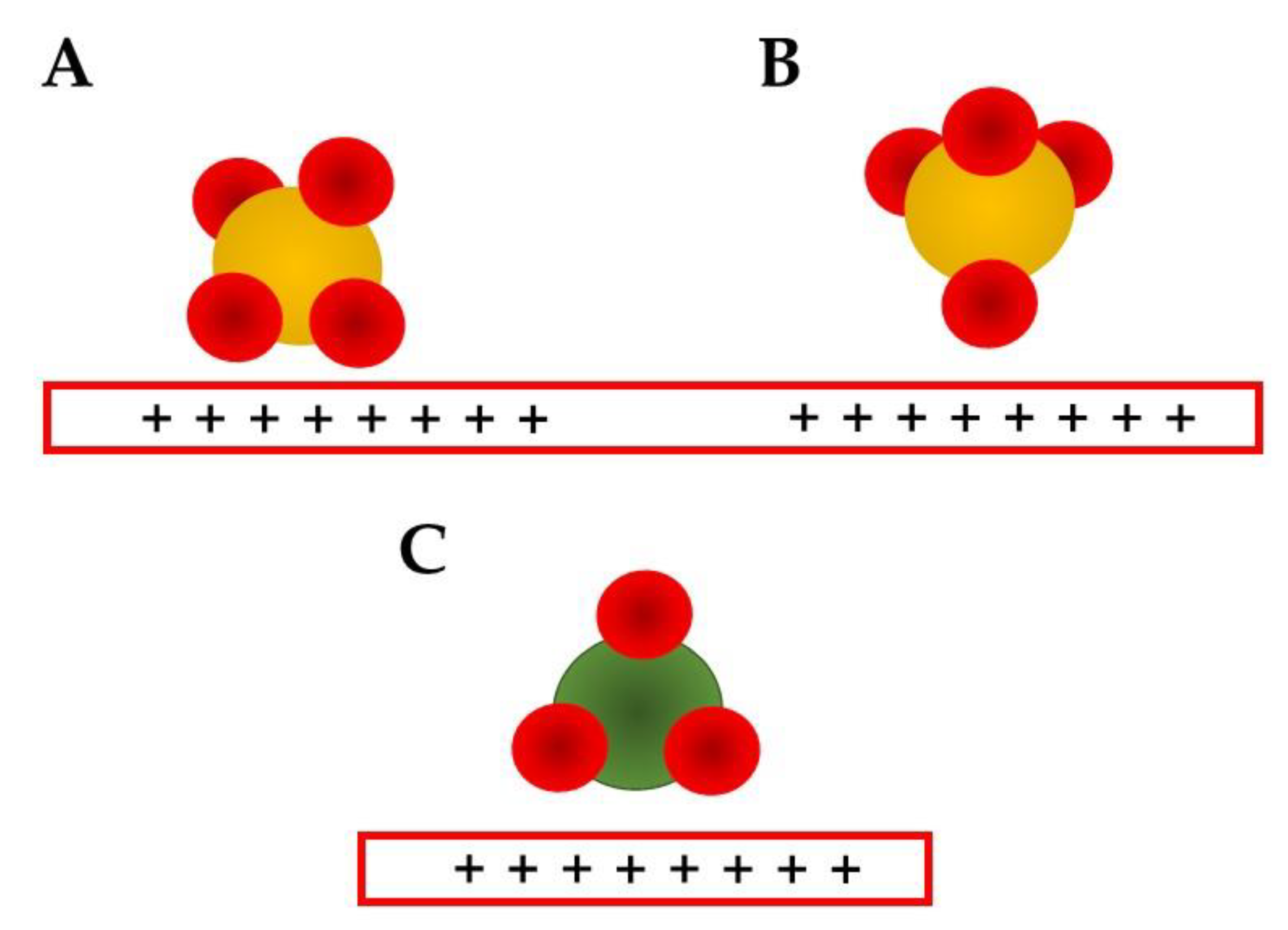
Figure 3. Proposed anion adsorption conformations for sulfate (A,B) and nitrate (C) [18].
For chaotropic anions, such as perchlorate and nitrate, the adsorption is not so strong. Especially for perchlorate, which is normally assumed to be a non-adsorbing anion [63][64][65]. However, there is already evidence that this anion is not just an adsorbate one [66][67][68][69], but that this adsorption also may present different conformations on the electrode surface [17]. The perchlorate adsorption is expected to present a duplicate of an asymmetric mode around 1250 cm−1 [17] and a symmetric mode stretching around 1100 cm−1 [7][8][9][17][28][29]. The symmetric one is reported to be more intense, and maybe this is the reason for it being mostly reported as the only perchlorate band. However, it is possible to find works that not only report both bands but also deconvolute the symmetric band in two, suggesting three moieties for the ClO4- adsorption [17].
Nitrate ions are reported to adsorb just by one conformation (Figure 3C) using two oxygen atoms and presenting just symmetric and asymmetric modes, at 1043 and 1440–1450 cm−1 [18] Halogenates, assumed in the Hofmeister series to be “neutral”, normally form chemical bonds with the electrode surface, and since they are monoatomic ions, the typical vibrations are M–X (M: metal, X: halogenate) stretching ones. These bands are observed at very low wavenumbers and, because of it, normally are in the region of absorption of the infrared cell window material, so they are observed just at Raman spectroscopy [32][57].
The presence of these ions in the EDL is reported to affect the electrochemical activity of several reactions, such as the SO2 oxidation reaction [28][29][51], and even some reduction reactions, such as oxygen reduction (ORR) [17]. The reduction reactions are normally observed at potentials more negative than the PZC, and because of it, the anions are expected to be repelled from the electrode surface. However, as shown by Nesselberger and Arenz, some anions, such as phosphate and sulfate, have a maximum surface coverage at potentials in which the ORR is already taking place [17]. In that work, the authors have shown that phosphoric acid adsorbs more and at more negative potentials than sulfate and/or perchlorate, suggesting a shift in PZC and a blocking of active sites at more negative potentials by phosphate anions.
The EDL can also present a different pH value than the bulk electrolyte [70][71][72][73][74][75][76]. Since hydronium and hydroxyl ions present vibrational bands, it is obvious that spectroelectrochemistry is also a powerful tool to investigate interfacial pH [27]. For more detailed information, the stretching of the hydronium complexes need also to be considered. The solvated hydronium, the Eigen cation (H3O+(H2O)2), has a very short lifetime, so unless the EDL is probed by a high-resolution spectroscopic technique, it will probably not be seen [77]. By contrast, the shared proton, Zundel complex (H2O…H+…H2O), is more stable [77] and possible to be observed in infrared spectroscopy applied to electrochemical systems by the band centered at 1700 cm−1 [7][8][9][28][29][56][78]. It also presents a characteristic stretching mode around 3160 cm−1 [77].
For obtaining more quantitative information about the local pH, the anions present in the electrolyte may be a more efficient probe. Ayemoba and Cuesta presented an interesting investigation on the interfacial pH variation during the CO2 reduction in Au electrodes [79] in which the intensity ratio between CO2 (2340 cm−1) and bicarbonate (1650 cm−1) related bands was used as a pH indicator based in the Henderson–Hasselbach equation. For such a study, the authors calibrated the system measuring this ratio using different solutions with bias-adjusted pH. The authors varied the cations in the electrolyte and investigated how they affect CO2 reduction. They have shown that for the biggest alkali cations, the ones with the lowest charge density and smaller solvation shells (the most chaotropic ones), the pH increment during the reaction is lower, and the electrochemical activity is higher [79].
Another anion pair that can be used as a pH probe is sulfate/bisulfate. Previous investigations have shown that with an increase in hydronium generation the sulfate-related band (1100 cm−1) decreases, while the symmetric (1000 cm−1) and asymmetric (1250 cm−1) bisulfate ones increase [54]. Investigating these bands, Dourado et al. [58] were able to calibrate an interfacial pH dependence on the bulk titrated pH and proposed a method to investigate the pKa of adsorbed species.
In alkaline media, the anion is hydroxyl (3750 cm−1) [59], and in a similar way as that explained for acidic media, this should not be the only ion presented in the EDL; the cations should also be present. However, these cations do not present vibrational bands by themselves, and normally no cation–electrode as well. In this specific case, it is suggested to further investigate the water-stretching region. In there, bands related to the solvated cations can be observed around 3630 cm−1, which are reported in the literature to be related to a Zundel-like complex involved in the solvation shell of sodium [7][10][80][81][82][83] or potassium ions [9][12][27].
Concepts to keep in mind:
-
Spectroelectrochemical techniques can provide the EDL structure evolution during an electrochemical process;
-
The water-related bands can be deconvoluted and different conformations can be obtained and investigated by them;
-
The potential shift of these water bands needs to be taken with care since they can be related to a generation or consumption behavior as well as a Stark shift. Simultaneous consideration of stretching and bending modes is mandatory;
-
Anions can also be followed by spectroscopic techniques, and their adsorption can be related to active sites blocking;
-
Cations are considered in alkaline media, but due to the lack of natural bands, the water in the solvation shell is responsible for the probing bands.
References
- Huang, J.; Chen, Y. Editorial Overview Surface electrochemistry (2022) The double layer: A persisting issue with emerging trends. Curr. Opin. Electrochem. 2022, 35, 101099.
- Schmickler, W. Double Layer Theory. J. Solid State Electrochem. 2020, 24, 2175–2176.
- Fawcett, W.R. Fifty Years of Studies of Double Layer Effects in Electrode Kinetics—A Personal View. J. Solid State Electrochem. 2011, 15, 1347–1358.
- Jacob, T.; Sabo, L. Electrochemical Double Layer Modeling Electrified Interfaces; Elsevier: Amsterdam, The Netherlands, 2018.
- Haid, R.W.; Ding, X.; Sarpey, T.K.; Bandarenka, A.S.; Garlyyev, B. Exploration of the Electrical Double-Layer Structure: Influence of Electrolyte Components on the Double-Layer Capacitance and Potential of Maximum Entropy. Curr. Opin. Electrochem. 2022, 32, 100882.
- Stamenkovic, V.R.; Strmcnik, D.; Lopes, P.P.; Markovic, N.M. Energy and Fuels from Electrochemical Interfaces. Nat. Mater. 2016, 16, 57–69.
- Ataka, K.; Yotsuyanagi, T.; Osawa, M. Potential-Dependent Reorientation of Water Molecules at an Electrode/Electrolyte Interface Studied by Surface-Enhanced Infrared Absorption Spectroscopy. J. Phys. Chem. 1996, 100, 10664–10672.
- Garcia-Araez, N.; Rodriguez, P.; Navarro, V.; Bakker, H.J.; Koper, M.T.M. Structural Effects on Water Adsorption on Gold Electrodes. J. Phys. Chem. C 2011, 115, 21249–21257.
- Garcia-Araez, N.; Rodriguez, P.; Bakker, H.J.; Koper, M.T.M. Effect of the Surface Structure of Gold Electrodes on the Coadsorption of Water and Anions. J. Phys. Chem. C 2012, 116, 4786–4792.
- Wandlowski, T.; Ataka, K.; Pronkin, S.; Diesing, D. Surface Enhanced Infrared Spectroscopy—Au(1 1 1–20 Nm)/Sulphuric Acid—New Aspects and Challenges. Electrochim. Acta 2004, 49, 1233–1247.
- Li, C.-Y.; Le, J.-B.; Wang, Y.-H.; Chen, S.; Yang, Z.-L.; Li, J.-F.; Cheng, J.; Tian, Z.-Q. In Situ Probing Electrified Interfacial Water Structures at Atomically Flat Surfaces. Nat. Mater. 2019, 18, 697–701.
- Chen, X.; McCrum, I.T.; Schwarz, K.A.; Janik, M.J.; Koper, M.T.M. Co-Adsorption of Cations as the Cause of the Apparent PH Dependence of Hydrogen Adsorption on a Stepped Platinum Single-Crystal Electrode. Angew. Chem. Int. Ed. 2017, 56, 15025–15029.
- Velasco-Velez, J.J.; Wu, C.H.; Pascal, T.A.; Wan, L.F.; Guo, J.; Prendergast, D.; Salmeron, M. The Structure of Interfacial Water on Gold Electrodes Studied by X-ray Absorption Spectroscopy. Science 2014, 346, 831–834.
- Favaro, M.; Jeong, B.; Ross, P.N.; Yano, J.; Hussain, Z.; Liu, Z.; Crumlin, E.J. Unravelling the Electrochemical Double Layer by Direct Probing of the Solid/Liquid Interface. Nat. Commun. 2016, 7, 12695.
- Shin, S.; Kim, D.H.; Ringe, S. Electric Double Layer Structure in Aqueous Electrolyte and Its Electrocatalytic Importance. Research Square, 2021; preprint.
- Ledezma-Yanez, I.; Wallace, W.D.Z.; Sebastián-Pascual, P.; Climent, V.; Feliu, J.M.; Koper, M.T.M. Interfacial Water Reorganization as a PH-Dependent Descriptor of the Hydrogen Evolution Rate on Platinum Electrodes. Nat. Energy 2017, 2, 17031.
- Nesselberger, M.; Arenz, M. In Situ FTIR Spectroscopy: Probing the Electrochemical Interface during the Oxygen Reduction Reaction on a Commercial Platinum High-Surface-Area Catalyst. ChemCatChem 2016, 8, 1125–1131.
- Moraes, I.R.; da Cunha, M.C.P.M.; Nart, F.C. Vibrational Spectroscopy of Adsorbed Sulfate and Nitrate Ions on Au(100) Electrodes. J. Braz. Chem. Soc. 1996, 7, 453–460.
- Martins, V.L.; Neves, H.R.; Monje, I.E.; Leite, M.M.; de Oliveira, P.F.M.; Antoniassi, R.M.; Chauque, S.; Morais, W.G.; Melo, E.C.; Obana, T.T.; et al. An Overview on the Development of Electrochemical Capacitors and Batteries—Part I. An. Acad. Bras. Cienc. 2020, 92, 1–28.
- Sagadevan, S.; Marlinda, A.R.; Chowdhury, Z.Z.; Wahab, Y.B.A.; Hamizi, N.A.; Shahid, M.M.; Mohammad, F.; Podder, J.; Johan, M.R. Fundamental Electrochemical Energy Storage Systems. In Advances in Supercapacitor and Supercapattery: Innovations in Energy Storage Devices; Elsevier: Amsterdam, The Netherlands, 2020; pp. 27–43. ISBN 9780128198971.
- Sagadevan, S.; Johan, M.R.; Marlinda, A.R.; Akbarzadeh, O.; Pandian, K.; Shahid, M.M.; Mohammad, F.; Podder, J. Background of Energy Storage. In Advances in Supercapacitor and Supercapattery: Innovations in Energy Storage Devices; Elsevier: Amsterdam, The Netherlands, 2020; pp. 1–26. ISBN 9780128198971.
- Bashir, S.; Chong, M.Y.; Hina, M.; Kamran, K.; Ramesh, S.; Ramesh, K. Aqueous Solid and Gel Electrolytes for Supercapattery. In Advances in Supercapacitor and Supercapattery: Innovations in Energy Storage Devices; Elsevier: Amsterdam, The Netherlands, 2020; pp. 271–310. ISBN 9780128198971.
- Wu, J. Understanding the Electric Double-Layer Structure, Capacitance, and Charging Dynamics. Chem. Rev. 2022, 122, 10821–10859.
- Drab, M.; Kralj-Iglič, V. Electric Double Layer of Electrons: Attraction between Two like-Charged Surfaces Induced by Fermi–Dirac Statistics. Phys. Lett. A 2019, 383, 358–365.
- Drab, M.; Kralj-Iglič, V. Diffuse Electric Double Layer in Planar Nanostructures Due to Fermi-Dirac Statistics. Electrochim. Acta 2016, 204, 154–159.
- Du, H.; Lin, X.; Xu, Z.; Chu, D. Electric Double-Layer Transistors: A Review of Recent Progress. J. Mater. Sci. 2015, 50, 5641–5673.
- De Rodrigues, M.P.S.; Dourado, A.H.B.; de Cutolo, L.O.; Parreira, L.S.; Alves, T.V.; Slater, T.J.A.; Haigh, S.J.; Camargo, P.H.C.; Cordoba de Torresi, S.I. Gold–Rhodium Nanoflowers for the Plasmon-Enhanced Hydrogen Evolution Reaction under Visible Light. ACS Catal. 2021, 11, 13543–13555.
- Dourado, A.H.B.; Silva, N.A.; Munhos, R.L.; del Colle, V.; Arenz, M.; Varela, H.; Córdoba de Torresi, S.I. Influence of Anion Chaotropicity on the SO2 Oxidation Reaction: When Spectator Species Determine the Reaction Pathway. ChemElectroChem 2020, 7, 1843–1850.
- Dourado, A.H.B.; Munhos, R.L.; Silva, N.A.; del Colle, V.; Carvalho, G.G.A.; Oliveira, P.V.; Arenz, M.; Varela, H.; Córdoba de Torresi, S.I. Opportunities and Knowledge Gaps of SO2 Electrocatalytic Oxidation for H2 Electrochemical Generation. ACS Catal. 2019, 9, 8136–8143.
- Lamoureux, P.S.; Singh, A.R.; Chan, K. PH Effects on Hydrogen Evolution and Oxidation over Pt(111): Insights from First-Principles. ACS Catal. 2019, 9, 6194–6201.
- Zheng, Y.; Jiao, Y.; Vasileff, A.; Qiao, S.Z. The Hydrogen Evolution Reaction in Alkaline Solution: From Theory, Single Crystal Models, to Practical Electrocatalysts. Angew. Chem. Int. Ed. 2018, 57, 7568–7579.
- Brolo, A.G.; Germain, P.; Hager, G. Investigation of the Adsorption of L-Cysteine on a Polycrystalline Silver Electrode by Surface-Enhanced Raman Scattering (SERS) and Surface-Enhanced Second Harmonic Generation (SESHG). J. Phys. Chem. B 2002, 106, 5982–5987.
- Mendes, R.K.; Freire, R.S.; Fonseca, C.P.; Neves, S.; Kubota, L.T. Characterization of Self-Assembled Thiols Monolayers on Gold Surface by Electrochemical Impedance Spectroscopy. J. Braz. Chem. Soc. 2004, 15, 849–855.
- Du, H.; Lin, X.; Xu, Z.; Chu, D. A Review of Molecular Modelling of Electric Double Layer Capacitors. Phys. Chem. Chem. Phys. 2014, 16, 6519–6538.
- Björneholm, O.; Hansen, M.H.; Hodgson, A.; Liu, L.M.; Limmer, D.T.; Michaelides, A.; Pedevilla, P.; Rossmeisl, J.; Shen, H.; Tocci, G.; et al. Water at Interfaces. Chem. Rev. 2016, 116, 7698–7726.
- Jensen, K.D.; Tymoczko, J.; Rossmeisl, J.; Bandarenka, A.; Chorkendorff, I.; Escudero-Escribano, M.; Stephens, I.E.L. Elucidation of the Oxygen Reduction Volcano in Alkaline Media Using a Copper-Platinum(111) Alloy. Angew. Chem. Int. Ed. 2018, 57, 2800–2805.
- Zana, A.; Wiberg, G.K.H.; Deng, Y.; Østergaard, T.; Rossmeisl, J.; Arenz, M. Accessing the Inaccessible: Analyzing the Oxygen Reduction Reaction in the Diffusion Limit. ACS Appl. Mater. Interfaces 2017, 9, 38176–38180.
- Sebastián-Pascual, P.; Petersen, A.S.; Bagger, A.; Rossmeisl, J.; Escudero-Escribano, M. PH and Anion Effects on Cu–Phosphate Interfaces for CO Electroreduction. ACS Catal. 2021, 1128–1135.
- Rossmeisl, J.; Chan, K.; Skúlason, E.; Björketun, M.E.; Tripkovic, V. On the PH Dependence of Electrochemical Proton Transfer Barriers. Catal. Today 2016, 262, 36–40.
- Tripkovic, V.; Björketun, M.E.; Skúlason, E.; Rossmeisl, J. Standard Hydrogen Electrode and Potential of Zero Charge in Density Functional Calculations. Phys. Rev. B Condens. Matter Mater. Phys. 2011, 84, 115452.
- Timoshenko, J.; Roldan Cuenya, B. In Situ/Operando Electrocatalyst Characterization by X-ray Absorption Spectroscopy. Chem. Rev. 2021, 121, 882–961.
- Mesa, C.A.; Pastor, E.; Francàs, L. UV-Vis Operando Spectroelectrochemistry for (Photo)Electrocatalysis: Principles and Guidelines. Curr. Opin. Electrochem. 2022, 35, 101098.
- Cuesta, A. ATR-SEIRAS for Time-Resolved Studies of Electrode–Electrolyte Interfaces. Curr. Opin. Electrochem. 2022, 35, 101041.
- Yu, A.; Chen, W.; Chen, Y.-X.; Zhu, B.-Q.; Sun, Z.-J.; Cai, J. Probing Complex Electrocatalytic Reactions Using Electrochemical in Situ Infrared Spectroscopy. Curr. Opin. Electrochem. 2019, 14, 113–123.
- Wang, H.; Zhou, Y.-W.; Cai, W.-B. Recent Applications of in Situ ATR-IR Spectroscopy in Interfacial Electrochemistry. Curr. Opin. Electrochem. 2017, 1, 73–79.
- Sanghapi, A.; Ramakrishan, S.; Fan, S.; Shannon, C. InSitu Characterization of Bipolar Electrodes by Using Vibrational Stark Spectroscopy and Surface-Enhanced Raman Spectroelectrochemistry. ChemElectroChem 2016, 3, 436–440.
- Kaim, W.; Fiedler, J. Spectroelectrochemistry: The Best of Two Worlds. Chem. Soc. Rev. 2009, 38, 3373–3382.
- Scherson, D.A.; Tolmachev, Y.V.; Stefan, I.C. Ultraviolet/Visible Spectroelectrochemistry; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2006; pp. 10172–10225.
- Dourado, A.H.B.; Arenz, M.; Córdoba de Torresi, S.I. Mechanism of Electrochemical L-Cysteine Oxidation on Pt. ChemElectroChem 2019, 6, 1009–1013.
- Blizanac, B.B.; Lucas, C.A.; Gallagher, M.E.; Arenz, M.; Ross, P.N.; Marković, N.M. Anion Adsorption, CO Oxidation, and Oxygen Reduction Reaction on a Au(100) Surface: The PH Effect. J. Phys. Chem. B 2004, 108, 625–634.
- Dourado, A.H.B.; del Colle, V.; Munhos, R.L.; Feliu, J.M.; Varela, H.; de Torresi, S.I.C. SO2 Electrooxidation Reaction on Pt Single Crystal Surfaces in Acidic Media: Electrochemical and in Situ FTIR Studies. Electrochim. Acta 2022, 403, 139601.
- Dourado, A.H.B.; da Silva, A.G.M.; Pastrián, F.A.C.; Munhos, R.L.; de Lima Batista, A.P.; de Oliveira-Filho, A.G.S.; Quiroz, J.; de Oliveira, D.C.; Camargo, P.H.C.; Córdoba de Torresi, S.I. In Situ FTIR Insights into the Electrooxidation Mechanism of Glucose as a Function of the Surface Facets of Cu2O-Based Electrocatalytic Sensors. J. Catal. 2019, 375, 95–103.
- Pastrián, F.A.C.; da Silva, A.G.M.; Dourado, A.H.B.; de Lima Batista, A.P.; de Oliveira-Filho, A.G.S.; Quiroz, J.; de Oliveira, D.C.; Camargo, P.H.C.; Córdoba de Torresi, S.I. Why Could the Nature of Surface Facets Lead to Differences in the Activity and Stability of Cu2O-Based Electrocatalytic Sensors? ACS Catal. 2018, 8, 6265–6272.
- Dourado, A.H.B.; de Lima Batista, A.P.; Oliveira-Filho, A.G.S.; Sumodjo, P.T.A.; Cordoba de Torresi, S.I. L-Cysteine Electrooxidation in Alkaline and Acidic Media: A Combined Spectroelectrochemical and Computational Study. RSC Adv. 2017, 7, 7492–7501.
- Nakamura, M.; Kato, H.; Hoshi, N. Infrared Spectroscopy of Water Adsorbed on M(111) (M = Pt, Pd, Rh, Au, Cu) Electrodes in Sulfuric Acid Solution. J. Phys. Chem. C 2008, 112, 9458–9463.
- Osawa, M.; Tsushima, M.; Mogami, H.; Samjeské, G.; Yamakata, A. Structure of Water at the Electrified Platinum-Water Interface: A Study by Surface-Enhanced Infrared Absorption Spectroscopy. J. Phys. Chem. C 2008, 112, 4248–4256.
- Shen, A.; Pemberton, J.E. Investigation of Trace Interfacial Water at Silver Electrodes in a Series of Normal Alcohols Using Surface Enhanced Raman Scattering. Phys. Chem. Chem. Phys. 1999, 1, 5677–5684.
- Dourado, A.H.B.; Silva, R.A.; Torresi, R.M.; Sumodjo, P.T.A.; Arenz, M.; Cordoba de Torresi, S.I. Kinetics, Assembling, and Conformation Control of L-Cysteine Adsorption on Pt Investigated by in Situ FTIR Spectroscopy and QCM-D. ChemPhysChem 2018, 19, 2340–2348.
- Brooker, J.; Christensen, P.A.; Hamnett, A.; He, R.; Paliteiro, C.A. Combined Scanning Tunnelling Microscopy and in Situ Fourier Transform Infrared Study of Dioxygen Reduction on Gold. Faraday Discuss. 1992, 94, 339.
- Doneux, T.; Buess-Herman, C.; Lipkowski, J. Electrochemical and FTIR Characterization of the Self-Assembled Monolayer of 2-Mercaptobenzimidazole on Au(111). J. Electroanal. Chem. 2004, 564, 65–75.
- Zinola, C.F.; Rodríguez, J.L.; Arévalo, M.C.; Pastor, E. FTIR Studies of Tyrosine Oxidation at Polycrystalline Pt and Pt(111) Electrodes. J. Electroanal. Chem. 2005, 585, 230–239.
- Ogura, K.; Kobayashi, M.; Nakayama, M.; Miho, Y. In-Situ FTIR Studies on the Electrochemical Oxidation of Histidine and Tyrosine. J. Electroanal. Chem. 1999, 463, 218–223.
- Ninham, B.W.; Duignan, T.T.; Parsons, D.F. Approaches to Hydration, Old and New: Insights through Hofmeister Effects. Curr. Opin. Colloid Interface Sci. 2011, 16, 612–617.
- Schwierz, N.; Horinek, D.; Sivan, U.; Netz, R.R. Reversed Hofmeister Series—The Rule Rather than the Exception. Curr. Opin. Colloid Interface Sci. 2016, 23, 10–18.
- Gibb, B.C. Hofmeister’s Curse. Nat. Chem. 2019, 11, 963–965.
- Marković, N.M.; Adžić, R.R.; Cahan, B.D.; Yeager, E.B. Structural Effects in Electrocatalysis: Oxygen Reduction on Platinum Low Index Single-Crystal Surfaces in Perchloric Acid Solutions. J. Electroanal. Chem. 1994, 377, 249–259.
- Attard, G.A.; Brew, A.; Hunter, K.; Sharman, J.; Wright, E. Specific Adsorption of Perchlorate Anions on Pt Single Crystal Electrodes. Phys. Chem. Chem. Phys. 2014, 16, 13689–13698.
- Gubanova, E.; Schmidt, T.O.; Watzele, S.; Alexandrov, V.; Bandarenka, A.S. Structure-Dependent Electrical Double-Layer Capacitances of the Basal Plane Pd(Hkl) Electrodes in HClO4. J. Phys. Chem. C 2022, 126, 11414–11420.
- Gómez-Marín, A.M.; Feliu, J.M. Pt(111) Surface Disorder Kinetics in Perchloric Acid Solutions and the Influence of Specific Anion Adsorption. Electrochim. Acta 2012, 82, 558–569.
- Wu, J.; Zheng, W.; Chen, Y. Factors Affecting the Cathode/Electrolyte Interfacial PH Change during Water Reduction: A Simulation Study. Int. J. Hydrogen Energy 2022, 47, 18597–18605.
- Monteiro, M.C.O.; Koper, M.T.M. Measuring Local PH in Electrochemistry. Curr. Opin. Electrochem. 2021, 25, 100649.
- Henckel, D.A.; Counihan, M.J.; Holmes, H.E.; Chen, X.; Nwabara, U.O.; Verma, S.; Rodríguez-López, J.; Kenis, P.J.A.; Gewirth, A.A. Potential Dependence of the Local PH in a CO2 Reduction Electrolyzer. ACS Catal. 2021, 11, 255–263.
- Jong, M.; Sleegers, N.; Schram, J.; Daems, D.; Florea, A.; de Wael, K. A Benzocaine-Induced Local Near-Surface PH Effect: Influence on the Accuracy of Voltammetric Cocaine Detection. Anal. Sens. 2021, 1, 54–62.
- Varela, A.S.; Kroschel, M.; Reier, T.; Strasser, P. Controlling the Selectivity of CO2 electroreduction on Copper: The Effect of the Electrolyte Concentration and the Importance of the Local PH. Catal. Today 2016, 260, 8–13.
- Wang, X.; Xu, C.; Jaroniec, M.; Zheng, Y.; Qiao, S.Z. Anomalous Hydrogen Evolution Behavior in High-PH Environment Induced by Locally Generated Hydronium Ions. Nat. Commun. 2019, 10, 4876.
- Zülke, A.; Perroni, P.; Machado, E.G.; Varela, H. Rrde Studies of Glycerol Electro-Oxidation: Local PH Variation and Oscillatory Dynamics. ECS Trans. 2017, 77, 1643–1650.
- Agmon, N. Infrared Spectroscopy: The Acid Test for Water Structure. Nat. Chem. 2016, 8, 206–207.
- Max, J.-J.; Trudel, M.; Chapados, C. Substraction of the Water Spectra from the Infrared Spectrum of Acidic and Alkaline Solutions. Appl. Spectrosc. 1998, 52, 963–969.
- Ayemoba, O.; Cuesta, A. Spectroscopic Evidence of Size-Dependent Buffering of Interfacial PH by Cation Hydrolysis during CO2 Electroreduction. ACS Appl. Mater. Interfaces 2017, 9, 27377–27382.
- Thamer, M.; de Marco, L.; Ramasesha, K.; Mandal, A.; Tokmakoff, A. Ultrafast 2D IR Spectroscopy of the Excess Proton in Liquid Water. Science 2015, 350, 78–82.
- Max, J.J.; Trudel, M.; Chapados, C. Infrared Titration of Aqueous Glycine. Appl. Spectrosc. 1998, 52, 226–233.
- Max, J.-J.; Ménichelli, C.; Chapados, C. Infrared Titration of Aqueous Sulfuric Acid. J. Phys. Chem. A 2000, 104, 2845–2858.
- Max, J.-J.; Chapados, C. Infrared Titration of Aqueous NaOH by Aqueous HCl. Can. J. Chem. 2000, 78, 64–72.
More
Information
Subjects:
Electrochemistry; Chemistry, Physical; Spectroscopy
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.7K
Revisions:
2 times
(View History)
Update Date:
09 Dec 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No