 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Kazuhiro Mio | -- | 2626 | 2022-12-08 12:18:58 | | | |
| 2 | Rita Xu | + 7 word(s) | 2633 | 2022-12-09 02:50:04 | | | | |
| 3 | Kazuhiro Mio | Meta information modification | 2633 | 2022-12-09 10:24:42 | | |
Video Upload Options
Membrane proteins play important roles in biological functions, with accompanying allosteric structure changes. Understanding intramolecular dynamics helps elucidate catalytic mechanisms and develop new drugs. In contrast to the various technologies for structural analysis, methods for analyzing intramolecular dynamics are limited. Single-molecule measurements using optical microscopy have been widely used for kinetic analysis.
1. Introduction
2. Measurement of Observing Dynamics of Membrane Protein
2.1. Single Molecular Dynamics Using Optical Measurement
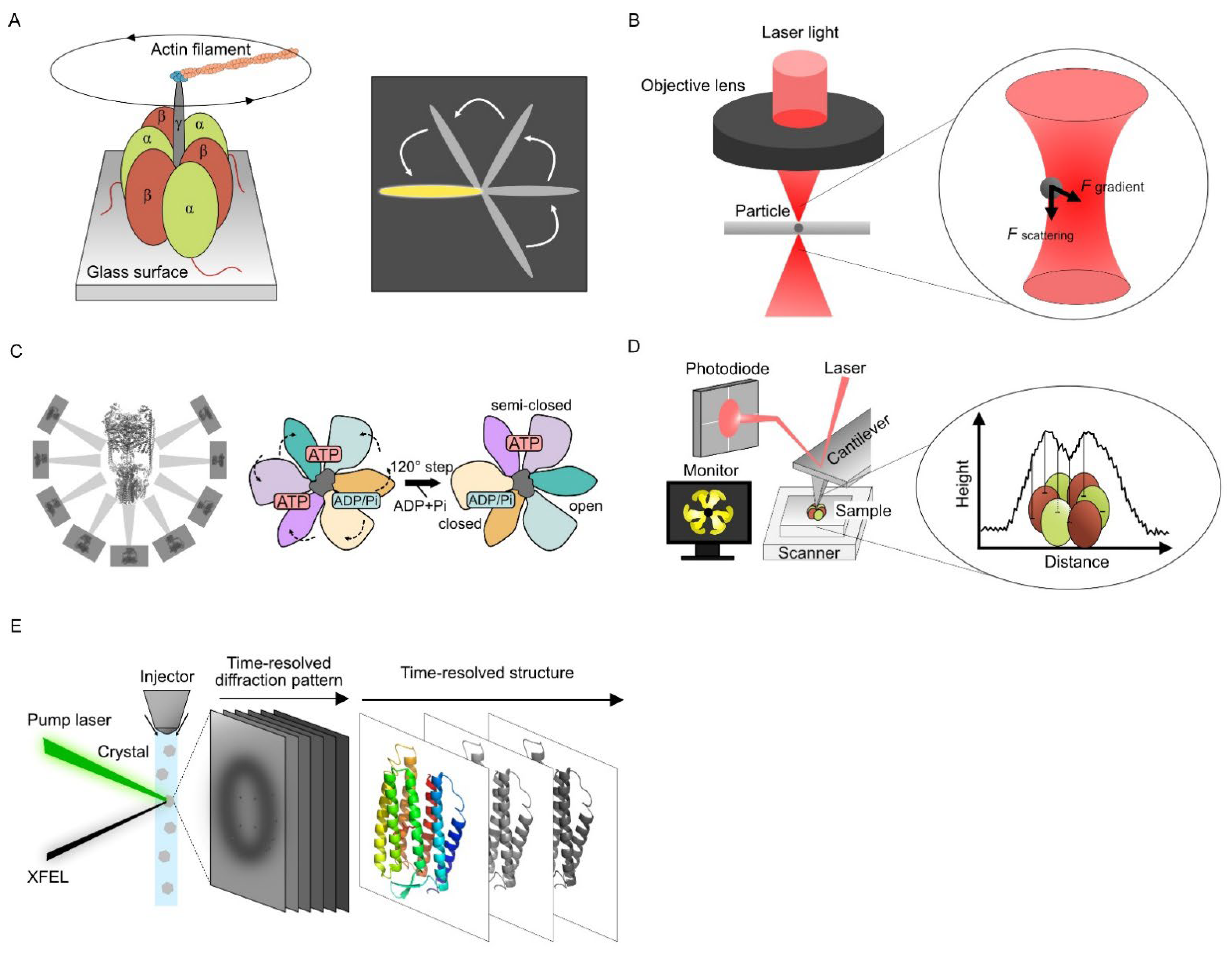
2.2. Single Particle Analysis Using Cryo-Electron Microscopy
2.3. Visualizing Protein Dynamics Using High-Speed Atomic Force Microscopy
HS-AFM images the structure of the sample surface by scanning with a sharp tip attached to the free end of a microcantilever and directly observing the dynamics and conformational information of the target protein simultaneously. Unlike steady-state conformations obtained from X-ray crystallography or cryo-EM, HS-AFM can observe the dynamics of raw sample images in an aqueous solution.
Ando et al. increased the scan speed of the AFM for imaging proteins, and the HS-AFM technology was established (Figure 1D) [27][28]. Direct observation of the F1-ATPase stator ring revealed the rotating movement of the three β-subunits continuously despite the lack of a γ-subunit [29]. The proper design of all HS-AFM components like scan heads or optimized ultra-short cantilevers is crucial to achieving excellent lateral and temporal resolution [30]. The prototyping and microfabrication of customized cantilevers are required to achieve fast data acquisition times (t), which is expressed as follows [31]:
where fc is the cantilever resonant frequency and N is the number of pixels per line.
HS-AFM has also been applied to living organisms. Yamashita et al. successfully observed dynamic molecular architectures on the surface of living cells [32], and Fukuma et al. developed 3D-AFM and imaged cell surfaces and intracellular structures in three dimensions using a long needle-like nanoprobe [33]. Combined measurements of HS-AFM with an optical microscope or 3D-AFM were also undertaken.
2.4. Time-Resolved Serial Femtosecond Crystallography Using X-ray Free Electron Laser
XFEL was developed as a new commercial X-ray light source in this century. In contrast to conventional synchrotron X-ray light sources, XFEL uses high-intensity and coherently amplified X-ray photons. In conventional X-ray crystal structure analysis, protein crystals are exposed to X-rays for several hundred milliseconds, inevitably causing structural damage to the crystal sample. XFEL uses subpicosecond- to picosecond-order pulsed X-rays, which are shorter than atomic movement after irradiation. Structural information can be obtained by XFEL before irradiation destroys the structure [34][35].
A combination of time-resolved serial femtosecond crystallography (TR-SFX) and the pump–probe techniques are widely used for XFEL dynamics analysis (Figure 1E). In TR-SFX, microcrystals are continuously fed into the XFEL-irradiated area using injectors [36], and the conformational changes of proteins within nanoseconds to milliseconds after laser irradiation can be observed as X-ray diffraction images. Time-resolved conformational changes can be captured by observing samples with different reaction times before X-ray irradiation. The photoexcitation kinetics of bacteriorhodopsin in nanoseconds to milliseconds were captured at the atomic level using TR-SFX [37]. The retinal isomerization-induced intramolecular dynamics and the involvement of water molecules even 10 µs after photoactivation were observed for the bacteriorhodopsin from its snapshots.
Pump-probe techniques can be used in time-resolved experiments using XFEL, especially for light-responsive proteins, including rhodopsin and a photosynthetic reaction center. In ligand-induced conformational dynamics, the mixing rate between protein crystals and a substrate determines their accuracy [38]. Developing a faster sample mixing system will enable time-resolved dynamics measurement of nonphotoresponsive membrane proteins, such as GPCRs, and ion channels.
2.5. Diffracted X-ray Tracking
DXT is a single-molecule technique using white X-rays, which can reveal the intramolecular dynamics of target proteins with a two-dimensional axis by tilting (θ) and twisting (χ) in an aqueous solution [39][40]. In DXT, the specific domain of the target proteins is labeled with gold nanocrystals, and the motions of the proteins are detected as the trajectories generated from the gold nanocrystals (Figure 2A). Intramolecular dynamics have been revealed using DXT, such as for DNA [40], a motor protein [41], and membrane proteins [42][43][44].
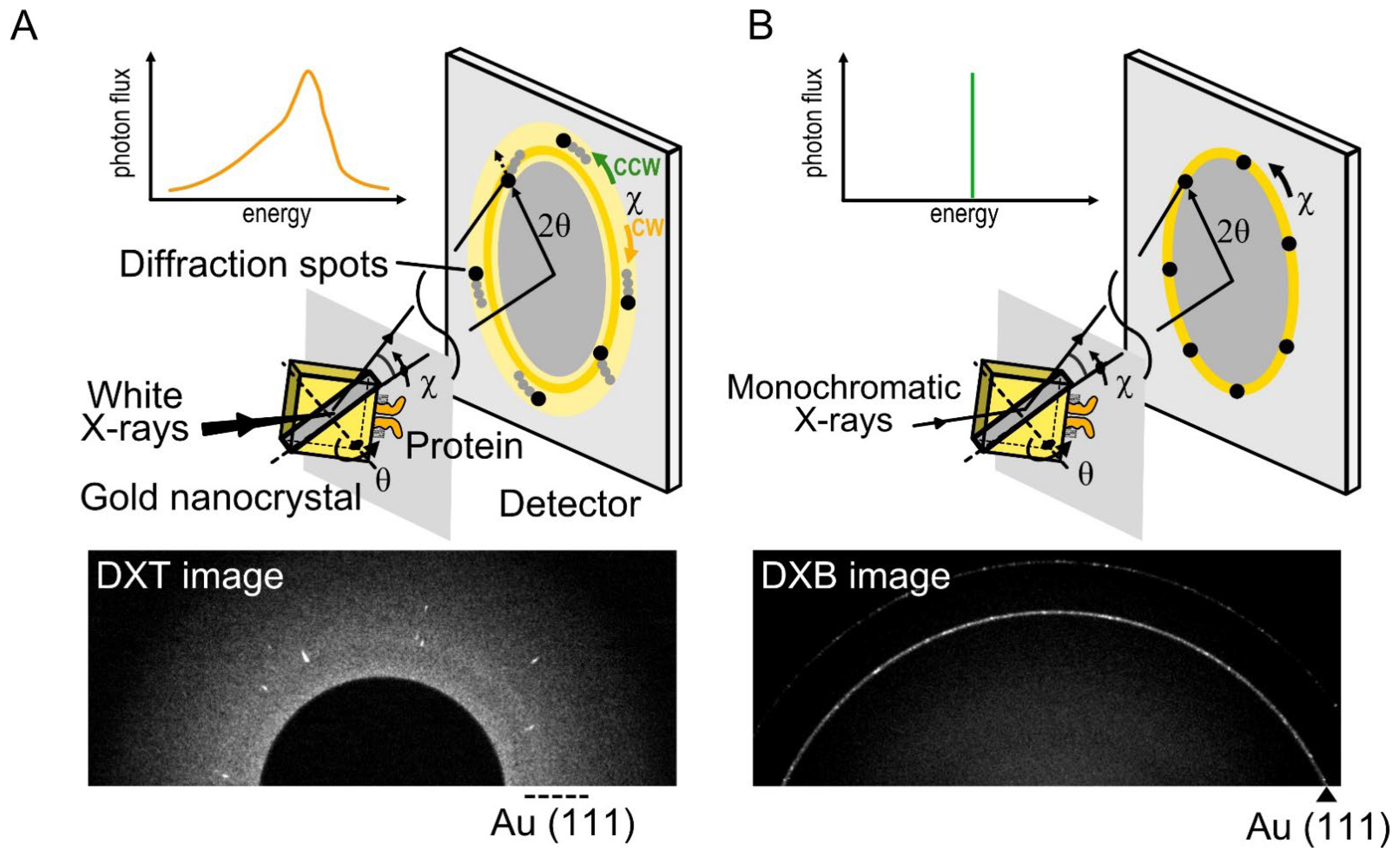
Figure 2. Diffracted X-ray tracking (DXT) and diffracted X-ray blinking (DXB) methods. (A) In DXT, the specific domain of a target protein is labeled with gold nanocrystals, with a diameter of 40 nm to 80 nm. The diffraction spots represent the tilting (θ) and twisting (χ) motions. DXT using white X-rays can analyze the trajectories of Laue spots appearing over a wide detector area. (B) DXB uses monochromatic X-rays, and the diffraction spots appear on the diffraction rings. The intramolecular motions are analyzed from the intensity changes of the Au (111) diffraction rings.
X-ray diffractions from gold nanocrystals are generated according to Bragg’s law as follows:
where n is the diffraction order, λ is the wavelength of the incident X-rays beam, d is the lattice plane spacing for a particular diffracting plane of atoms, and θ is the angle between the incident beam and the diffracting plane. DXT requires white X-rays with continuous wavelengths (energy widths ranging from 14.0 to 16.5 keV). Based on Bragg’s law, the angular displacement changes of the diffraction spots can be trackable.
There are three main advantages to DXT. First, DXT can track intramolecular movements with excellent spatiotemporal resolution. The spatial resolution of single-molecule measurements is expressed as λ/100, where λ is the wavelength. Because the X-rays wavelength used in DXT is 0.01 nm to 0.1 nm, positioning accuracy can be achieved at the picometer level, which is sufficient to recognize the intramolecular movements. In addition, DXT can measure dynamics with microsecond to millisecond order due to the high-brightness X-rays.
Second, DXT can obtain the dynamics data separately via the tilting (θ) and twisting (χ) motions. The motion of diffraction spots can be continuously tracked, and the detection limit is up to 1 mrad in the θ direction and 6 mrad in the χ direction. The motion of diffraction spots is analyzed using mean square displacement (MSD), the 2D histogram, and the mean plot of the angular displacements.
Third, DXT can be performed even under solution conditions. Target proteins are immobilized on a polyimide film substrate using chemically anchoring, antigen–antibody reactions, biotin–avidin reactions, or glycoprotein–lectin reactions. Then, gold nanocrystals are attached to the target domain of proteins using the Au–S reaction, the antigen–antibody reaction, etc. Samples in aqueous solution are sandwiched again with polyimide film to perform protein functions at maximum conditions. Here researchers present examples of dynamics analysis of the TRPV1 channel [44] and 5-HT2A serotonin receptor (5-HT2AR) [45][46]. In the case of DXT at the SPring-8 BL40XU beamline, diffraction images were recorded using an X-ray image intensifier (V5445P and V7739P, Hamamatsu Photonics, respectively) and a CMOS camera (FASTCAM SA1.1, Photron, Japan and Phantom V2511, Vision Research, respectively). Gold nanocrystals were deposited on the substrate surface of NaCl (100) or KCl (100) and grown epitaxially to a 40–80 nm diameter to obtain sufficient diffraction signals. The size of the gold nanocrystals is larger than that of proteins, and protein movement is affected by the attached gold nanocrystals in a size-dependent manner [43]. The obtained diffraction spots were tracked by the TrackPy program (v0.3.2 https://doi.org/10.5281/zenodo.60550) (accessed on 20 November 2022) after correcting the background. The trajectories were analyzed using IGOR Pro software (Wavemetrics, Lake Oswego).
The dynamics data obtained from DXT are restricted to movements in labeled domains of the proteins. Therefore, DXT techniques complement other two-dimensional or three-dimensional imaging techniques, such as cryo-EM, HS-AFM, and MD simulation. Combined analysis of these techniques provides the global dynamics of target protein molecules. In addition, DXT can choose suitable frame rates from microseconds to seconds. This means that the directly observed fast molecular motions can be complemented with atomic-level calculations from MD simulation analysis.
The methods for visualizing membrane protein dynamics are summarized in Table 1.
Table 1. Summary of methods for visualizing membrane protein dynamics.
|
Method |
Temporal resolution |
Spatial resolution |
Labeling |
Observation area |
Motion in solution |
|
Optical microscopy |
10 ms – 100 ms |
150 μm |
Florescent probe |
Target domain |
Possible |
|
Optical tweezers |
10 μs – min |
0.2 nm |
Beads attached |
Target domain |
Possible |
|
Cryo-electron |
None |
0.2 nm |
No label |
3D structure |
Impossible |
|
Atomic force |
30 s – 1 min |
1 nm |
No label |
Surface structure |
Impossible |
|
High-speed atomic force microscopy |
ms – s |
2 – 3 nm |
No label |
Surface structure |
Possible |
|
X-ray free electron |
fs – ps |
0.2 nm |
No label |
3D structure |
Impossible |
|
Diffracted X-ray |
ns – ms |
< 0.01 nm |
Gold nanocrystals |
Target domain |
Possible |
References
- Tran, N.T.; Mentink-Vigier, F.; Long, J.R. Dynamic Nuclear Polarization of Biomembrane Assemblies. Biomolecules 2020, 10, 1246.
- Bouvier, G.; Bardiaux, B.; Pellarin, R.; Rapisarda, C.; Nilges, M. Building Protein Atomic Models from Cryo-EM Density Maps and Residue Co-Evolution. Biomolecules 2022, 12, 1290.
- Sabbert, D.; Engelbrecht, S.; Junge, W. Intersubunit rotation in active F-ATPase. Nature 1996, 381, 623–625.
- Noji, H.; Yasuda, R.; Yoshida, M.; Kinosita, K., Jr. Direct observation of the rotation of F1-ATPase. Nature 1997, 386, 299–302.
- Boyer, P.D. The ATP synthase-a splendid molecular machine. Annu. Rev. Biochem. 1997, 66, 717–749.
- Kishikawa, J.; Nakanishi, A.; Nakano, A.; Saeki, S.; Furuta, A.; Kato, T.; Mistuoka, K.; Yokoyama, K. Structural snapshots of V/A-ATPase reveal the rotary catalytic mechanism of rotary ATPases. Nat. Commun. 2022, 13, 1213.
- Toprak, E.; Kural, C.; Selvin, P.R. Super-accuracy and super-resolution getting around the diffraction limit. Methods Enzymol. 2010, 475, 1–26.
- Ashkin, A.; Dziedzic, J.M.; Bjorkholm, J.E.; Chu, S. Observation of a single-beam gradient force optical trap for dielectric particles. Opt. Lett. 1986, 11, 288.
- Abbondanzieri, E.A.; Greenleaf, W.J.; Shaevitz, J.W.; Landick, R.; Block, S.M. Direct observation of base-pair stepping by RNA polymerase. Nature 2005, 438, 460–465.
- Sudhakar, S.; Abdosamadi, M.K.; Jachowski, T.J.; Bugiel, M.; Jannasch, A.; Schäffer, E. Germanium nanospheres for ultraresolution picotensiometry of kinesin motors. Science 2021, 371, 6530.
- Cecconi, C.; Shank, E.A.; Bustamante, C.; Marqusee, S. Direct observation of the three-state folding of a single protein molecule. Science 2005, 309, 2057–2060.
- Ma, L.; Cai, Y.; Li, Y.; Jiao, J.; Wu, Z.; O’Shaughnessy, B.; de Camilli, P.; Karatekin, E.; Zhang, Y. Single-molecule force spectroscopy of protein-membrane interactions. eLife 2017, 6, e30493.
- Pelz, B.; Žoldák, G.; Zeller, F.; Zacharias, M.; Rief, M. Subnanometre enzyme mechanics probed by single-molecule force spectroscopy. Nat. Commun. 2016, 7, 10848.
- Falleroni, F.; Bocchero, U.; Mortal, S.; Li, Y.; Ye, Z.; Cojoc, D.; Torre, V. Mechanotransduction in hippocampal neurons operates under localized low picoNewton forces. iScience 2022, 25, 103807.
- McMullan, G.; Faruqi, A.R.; Henderson, R. Direct Electron Detectors. Methods Enzymol. 2016, 579, 1–17.
- Brilot, A.F.; Chen, J.Z.; Cheng, A.; Pan, J.; Harrison, S.C.; Potter, C.S.; Carragher, B.; Henderson, R.; Grigorieff, N. Beam-induced motion of vitrified specimen on holey carbon film. J. Struct. Biol. 2012, 177, 630–637.
- Li, X.; Mooney, P.; Zheng, S.; Booth, C.R.; Braunfeld, M.B.; Gubbens, S.; Agard, D.A.; Cheng, Y. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat. Methods. 2013, 10, 584–590.
- Scheres, S.H. A Bayesian view on cryo-EM structure determination. J. Mol. Biol. 2012, 415, 406–418.
- Adrian, M.; Dubochet, J.; Lepault, J.; McDowall, A.W. Cryo-electron microscopy of viruses. Nature 1984, 308, 32–36.
- Dubochet, J.; Adrian, M.; Chang, J.J.; Homo, J.C.; Lepault, J.; McDowall, A.W.; Schultz, P. Cryo-electron microscopy of vitrified specimens. Q Rev. Biophys. 1988, 21, 129–228.
- Henderson, R.; Baldwin, J.M.; Ceska, T.A.; Zemlin, F.; Beckmann, E.; Downing, K.H. Model for the structure of bacteriorhodopsin based on high-resolution electron cryo-microscopy. J. Mol. Biol. 1990, 213, 899–929.
- Frank, J. Three-Dimensional Electron Microscopy of Macromolecular Assemblies; Academic Press: New York, NY, USA, 2006.
- Liao, M.; Cao, E.; Julius, D.; Cheng, Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 2013, 504, 107–112.
- Cao, E.; Liao, M.; Cheng, Y.; Julius, D.; TRPV1 structures in distinct conformations reveal activation mechanisms. Nature 2013, 504, 113–118.
- Nakanishi, A.; Kishikawa, J.I.; Tamakoshi, M.; Mitsuoka, K.; Yokoyama, K. Cryo-EM structure of intact rotary H+-ATPase/synthase from Thermus thermophilus. Nat. Commun. 2018, 9, 89.
- Doyle, M.T.; Jimah, J.R.; Dowdy, T.; Ohlemacher, S.I.; Larion, M.; Hinshaw, J.E.; Bernstein, H.D. Cryo-EM structures reveal multiple stages of bacterial outer membrane protein folding. Cell 2022, 185, 1143–1156.e13.
- Ando, T.; Kodera, N.; Takai, E.; Maruyama, D.; Saito, K.; Toda, A. A high-speed atomic force microscope for studying biological macromolecules. Proc. Natl. Acad. Sci. USA 2001, 98, 12468–12472.
- Ando, T.; Uchihashi, T.; Scheuring, S. Filming biomolecular processes by high-speed atomic force microscopy. Chem. Rev. 2014, 114, 3120–3188.
- Uchihashi, T.; Iino, R.; Ando, T.; Noji, H. High-speed atomic force microscopy reveals rotary catalysis of rotorless F₁-ATPase. Science 2011, 333, 755–758.
- Valotteau, C.; Sumbul, F.; Rico, F. High-speed force spectroscopy: Microsecond force measurements using ultrashort cantilevers. Biophys. Rev. 2019, 11, 689–699.
- Ando, T.; Kodera, N.; Naito, Y.; Kinoshita, T.; Furuta, K.; Toyoshima, Y.Y. A high-speed atomic force microscope for studying biological macromolecules in action. Chemphyschem 2003, 4, 1196–1202.
- Yamashita, H.; Taoka, A.; Uchihashi, T.; Asano, T.; Ando, T.; Fukumori, Y. Single-molecule imaging on living bacterial cell surface by high-speed AFM. J. Mol. Biol. 2012, 422, 300–309.
- Fukuma, T.; Ueda, Y.; Yoshioka, S.; Asakawa, H. Atomic-scale distribution of water molecules at the mica-water interface visualized by three-dimensional scanning force microscopy. Phys. Rev. Lett. 2010, 104, 016101.
- Neutze, R.; Wouts, R.; van der Spoel, D.; Weckert, E.; Hajdu, J. Potential for biomolecular imaging with femtosecond X-ray pulses. Nature 2000, 406, 752–757.
- Gaffney, K.J.; Chapman, H.N. Imaging atomic structure and dynamics with ultrafast x-ray scattering. Science 2007, 316, 1444–1448.
- Spence, J.C.; Doak, R.B. Single molecule diffraction. Phys. Rev. Lett. 2004, 92, 198102.
- Nango, E.; Royant, A.; Kubo, M.; Nakane, T.; Wickstrand, C.; Kimura, T.; Tanaka, T.; Tono, K.; Song, C.; Tanaka, R.; et al. A three-dimensional movie of structural changes in bacteriorhodopsin. Science 2016, 354, 1552–1557.
- Stagno, J.R.; Liu, Y.; Bhandari, Y.R.; Conrad, C.E.; Panja, S.; Swain, M.; Fan, L.; Nelson, G.; Li, C.; Wendel, D.R.; et al. Structures of riboswitch RNA reaction states by mix-and-inject XFEL serial crystallography. Nature 2017, 541, 242–246.
- Sasaki, Y.C.; Suzuki, Y.; Yagi, N.; Adachi, S.; Ishibashi, M.; Suda, H.; Toyota, K.; Yanagihara, M. Tracking of individual nanocrystals using diffracted x rays. Phys. Rev. E 2000, 62, 3843–3847.
- Sasaki, Y.C.; Okumura, Y.; Adachi, S.; Suda, H.; Taniguchi, Y.; Yagi, N. Picometer-scale dynamical x-ray imaging of single DNA molecules. Phys. Rev. Lett. 2001, 87, 248102.
- Sekiguchi, H.; Nakagawa, A.; Moriya, K.; Makabe, K.; Ichiyanagi, K.; Nozawa, S.; Sato, T.; Adachi, S.; Kuwajima, K.; Yohda, M.; et al. ATP dependent rotational motion of group II chaperonin observed by X-ray single molecule tracking. PLoS ONE 2013, 8, e64176.
- Shimizu, H.; Iwamoto, M.; Konno, T.; Nihei, A.; Sasaki, Y.C.; Oiki, S. Global twisting motion of single molecular KcsA potassium channel upon gating. Cell 2008, 132, 67–78.
- Sekiguchi, H.; Suzuki, Y.; Nishino, Y.; Kobayashi, S.; Shimoyama, Y.; Cai, W.; Nagata, K.; Okada, M.; Ichiyanagi, K.; Ohta, N.; et al. Real time ligand-induced motion mappings of AChBP and nAChR using X-ray single molecule tracking. Sci. Rep. 2014, 4, 6384.
- Fujimura, S.; Mio, K.; Kuramochi, M.; Sekiguchi, H.; Ikezaki, K.; Mio, M.; Hengphasatporn, K.; Shigeta, Y.; Kubo, T.; Sasaki, Y.C. Agonist and Antagonist-Diverted Twisting Motions of a Single TRPV1 Channel. J. Phys. Chem. B. 2020, 124, 11617–11624.
- Mio, K.; Ishihara, M.; Fujimura, S.; Sasaki, D.; Nozawa, S.; Ichiyanagi, K.; Fukaya, R.; Adachi, S.I.; Kuramochi, M.; Sekiguchi, H.; et al. X-ray-based living-cell motion analysis of individual serotonin receptors. Biochem. Biophys. Res. Commun. 2020, 529, 306–313.
- Mio, K.; Fujimura, S.; Ishihara, M.; Kuramochi, M.; Sekiguchi, H.; Kubo, T.; Sasaki, Y.C. Living-Cell Diffracted X-ray Tracking Analysis Confirmed Internal Salt Bridge Is Critical for Ligand-Induced Twisting Motion of Serotonin Receptors. Int. J. Mol. Sci. 2021, 22, 5285.

