 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Dean Liu | -- | 4395 | 2022-12-01 01:41:55 |
Video Upload Options
Catenin beta-1, also known as β-catenin, is a protein that in humans is encoded by the CTNNB1 gene. β-catenin is a dual function protein, involved in regulation and coordination of cell–cell adhesion and gene transcription. In humans, the CTNNB1 protein is encoded by the CTNNB1 gene. In Drosophila, the homologous protein is called armadillo. β-catenin is a subunit of the cadherin protein complex and acts as an intracellular signal transducer in the Wnt signaling pathway. It is a member of the catenin protein family and homologous to γ-catenin, also known as plakoglobin. Beta-catenin is widely expressed in many tissues. In cardiac muscle, beta-catenin localizes to adherens junctions in intercalated disc structures, which are critical for electrical and mechanical coupling between adjacent cardiomyocytes. Mutations and overexpression of β-catenin are associated with many cancers, including hepatocellular carcinoma, colorectal carcinoma, lung cancer, malignant breast tumors, ovarian and endometrial cancer. Alterations in the localization and expression levels of beta-catenin have been associated with various forms of heart disease, including dilated cardiomyopathy. β-catenin is regulated and destroyed by the beta-catenin destruction complex, and in particular by the adenomatous polyposis coli (APC) protein, encoded by the tumour-suppressing APC gene. Therefore, genetic mutation of the APC gene is also strongly linked to cancers, and in particular colorectal cancer resulting from familial adenomatous polyposis (FAP).
1. Discovery
Beta-catenin was initially discovered in the early 1990s as a component of a mammalian cell adhesion complex: a protein responsible for cytoplasmatic anchoring of cadherins.[1] But very soon, it was realized that the Drosophila protein armadillo – implicated in mediating the morphogenic effects of Wingless/Wnt – is homologous to the mammalian β-catenin, not just in structure but also in function.[2] Thus beta-catenin became one of the first examples of moonlighting: a protein performing more than one radically different cellular function.
2. Structure
2.1. Protein Structure
The core of beta-catenin consists of several very characteristic repeats, each approximately 40 amino acids long. Termed armadillo repeats, all these elements fold together into a single, rigid protein domain with an elongated shape – called armadillo (ARM) domain. An average armadillo repeat is composed of three alpha helices. The first repeat of β-catenin (near the N-terminus) is slightly different from the others – as it has an elongated helix with a kink, formed by the fusion of helices 1 and 2.[3] Due to the complex shape of individual repeats, the whole ARM domain is not a straight rod: it possesses a slight curvature, so that an outer (convex) and an inner (concave) surface is formed. This inner surface serves as a ligand-binding site for the various interaction partners of the ARM domains.
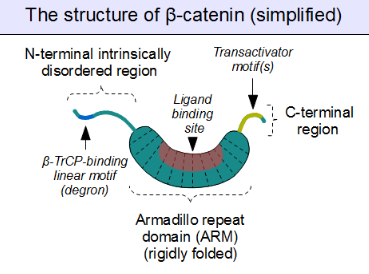
The segments N-terminal and far C-terminal to the ARM domain do not adopt any structure in solution by themselves. Yet these intrinsically disordered regions play a crucial role in beta-catenin function. The N-terminal disordered region contains a conserved short linear motif responsible for binding of TrCP1 (also known as β-TrCP) E3 ubiquitin ligase – but only when it is phosphorylated. Degradation of β-catenin is thus mediated by this N-terminal segment. The C-terminal region, on the other hand, is a strong transactivator when recruited onto DNA. This segment is not fully disordered: part of the C-terminal extension forms a stable helix that packs against the ARM domain, but may also engage separate binding partners.[4] This small structural element (HelixC) caps the C-terminal end of the ARM domain, shielding its hydrophobic residues. HelixC is not necessary for beta-catenin to function in cell–cell adhesion. On the other hand, it is required for Wnt signaling: possibly to recruit various coactivators, such as 14-3-3zeta.[5] Yet its exact partners among the general transcription complexes are still incompletely understood, and they likely involve tissue-specific players.[6] Notably, the C-terminal segment of β-catenin can mimic the effects of the entire Wnt pathway if artificially fused to the DNA binding domain of LEF1 transcription factor.[7]
Plakoglobin (also called gamma-catenin) has a strikingly similar architecture to that of beta-catenin. Not only their ARM domains resemble each other in both architecture and ligand binding capacity, but the N-terminal β-TrCP-binding motif is also conserved in plakoglobin, implying common ancestry and shared regulation with β-catenin.[8] However, plakoglobin is a very weak transactivator when bound to DNA – this is probably caused by the divergence of their C-terminal sequences (plakoglobin appears to lack the transactivator motifs, and thus inhibits the Wnt pathway target genes instead of activating them).[9]
2.2. Partners Binding to the Armadillo Domain

As sketched above, the ARM domain of beta-catenin acts as a platform to which specific linear motifs may bind. Located in structurally diverse partners, the β-catenin binding motifs are typically disordered on their own, and typically adopt a rigid structure upon ARM domain engagement – as seen for short linear motifs. However, β-catenin interacting motifs also have a number of peculiar characteristics. First, they might reach or even surpass the length of 30 amino acids in length, and contact the ARM domain on an excessively large surface area. Another unusual feature of these motifs is their frequently high degree of phosphorylation. Such Ser/Thr phosphorylation events greatly enhance the binding of many β-catenin associating motifs to the ARM domain.[10]
The structure of beta-catenin in complex with the catenin binding domain of the transcriptional transactivation partner TCF provided the initial structural roadmap of how many binding partners of beta-catenin may form interactions.[11] This structure demonstrated how the otherwise disordered N-terminus of TCF adapted what appeared to be a rigid conformation, with the binding motif spanning many beta-catenin repeats. Relatively strong charged interaction "hot spots" were defined (predicted, and later verified, to be conserved for the beta-catenin/E-cadherin interaction), as well as hydrophobic regions deemed important in the overall mode of binding and as potential therapeutic small molecule inhibitor targets against certain cancer forms. Furthermore, following studies demonstrated another peculiar characteristic, plasticity in the binding of the TCF N-terminus to beta-catenin.[12][13]
Similarly, we find the familiar E-cadherin, whose cytoplasmatic tail contacts the ARM domain in the same canonical fashion.[14] The scaffold protein axin (two closely related paralogs, axin 1 and axin 2) contains a similar interaction motif on its long, disordered middle segment.[15] Although one molecule of axin only contains a single β-catenin recruitment motif, its partner the Adenomatous Polyposis Coli (APC) protein contains 11 such motifs in tandem arrangement per protomer, thus capable to interact with several β-catenin molecules at once.[16] Since the surface of the ARM domain can typically accommodate only one peptide motif at any given time, all these proteins compete for the same cellular pool of β-catenin molecules. This competition is the key to understand how the Wnt signaling pathway works.
However, this "main" binding site on the ARM domain β-catenin is by no means the only one. The first helices of the ARM domain form an additional, special protein-protein interaction pocket: This can accommodate a helix-forming linear motif found in the coactivator BCL9 (or the closely related BCL9L) – an important protein involved in Wnt signaling.[17] Although the precise details are much less clear, it appears that the same site is used by alpha-catenin when beta-catenin is localized to the adherens junctions.[18] Because this pocket is distinct from the ARM domain's "main" binding site, there is no competition between alpha-catenin and E-cadherin or between TCF1 and BCL9, respectively.[19] On the other hand, BCL9 and BCL9L must compete with α-catenin to access β-catenin molecules.[20]
3. Function
3.1. Regulation of Degradation Through Phosphorylation
The cellular level of beta-catenin is mostly controlled by its ubiquitination and proteosomal degradation. The E3 ubiquitin ligase TrCP1 (also known as β-TrCP) can recognize β-catenin as its substrate through a short linear motif on the disordered N-terminus. However, this motif (Asp-Ser-Gly-Ile-His-Ser) of β-catenin needs to be phosphorylated on the two serines in order to be capable to bind β-TrCP. Phosphorylation of the motif is performed by Glycogen Synthase Kinase 3 alpha and beta (GSK3α and GSK3β). GSK3s are constitutively active enzymes implicated in several important regulatory processes. There is one requirement, though: substrates of GSK3 need to be pre-phosphorylated four amino acids downstream (C-terminally) of the actual target site. Thus it also requires a "priming kinase" for its activities. In the case of beta-catenin, the most important priming kinase is Casein Kinase I (CKI). Once a serin-threonine rich substrate has been "primed", GSK3 can "walk" across it from C-terminal to N-terminal direction, phosphorylating every 4th serine or threonine residues in a row. This process will result in dual phosphorylation of the aforementioned β-TrCP recognition motif as well.
3.2. The Beta-Catenin Destruction Complex
For GSK3 to be a highly effective kinase on a substrate, pre-phosphorylation is not enough. There is one additional requirement: Similar to the mitogen-activated protein kinases (MAPKs), substrates need to associate with this enzyme through high-affinity docking motifs. Beta-catenin contains no such motifs, but a special protein does: axin. What is more, its GSK3 docking motif is directly adjacent to a β-catenin binding motif.[15] This way, axin acts as a true scaffold protein, bringing an enzyme (GSK3) together with its substrate (β-catenin) into close physical proximity.
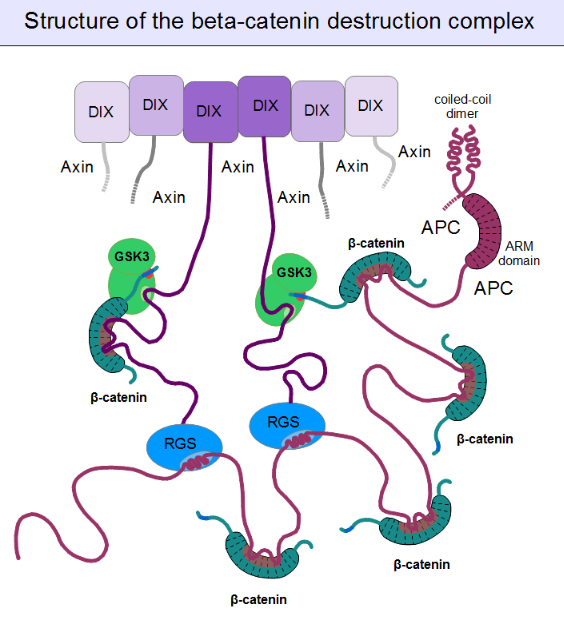
But even axin does not act alone. Through its N-terminal regulator of G-protein signaling (RGS) domain, it recruits the adenomatous polyposis coli (APC) protein. APC is like a huge "Christmas tree": with a multitude of β-catenin binding motifs (one APC molecule alone possesses 11 such motifs [16]), it may collect as many β-catenin molecules as possible.[21] APC can interact with multiple axin molecules at the same time as it has three SAMP motifs (Ser-Ala-Met-Pro) to bind the RGS domains found in axin. In addition, axin also has the potential to oligomerize through its C-terminal DIX domain. The result is a huge, multimeric protein assembly dedicated to β-catenin phosphorylation. This complex is usually called the beta-catenin destruction complex, although it is distinct from the proteosome machinery actually responsible for β-catenin degradation.[22] It only marks β-catenin molecules for subsequent destruction.
3.3. Wnt Signaling and the Regulation of Destruction
In resting cells, axin molecules oligomerize with each other through their C-terminal DIX domains, which have two binding interfaces. Thus they can build linear oligomers or even polymers inside the cytoplasm of cells. DIX domains are unique: the only other proteins known to have a DIX domain are Dishevelled and DIXDC1. (The single Dsh protein of Drosophila corresponds to three paralogous genes, Dvl1, Dvl2 and Dvl3 in mammals.) Dsh associates with the cytoplasmic regions of Frizzled receptors with its PDZ and DEP domains. When a Wnt molecule binds to Frizzled, it induces a poorly known cascade of events, that result in the exposure of dishevelled's DIX domain and the creation of a perfect binding site for axin. Axin is then titrated away from its oligomeric assemblies – the β-catenin destruction complex – by Dsh.[23] Once bound to the receptor complex, axin will be rendered incompetent for β-catenin binding and GSK3 activity. Importantly, the cytoplasmic segments of the Frizzled-associated LRP5 and LRP6 proteins contain GSK3 pseudo-substrate sequences (Pro-Pro-Pro-Ser-Pro-x-Ser), appropriately "primed" (pre-phosphorylated) by CKI, as if it were a true substrate of GSK3. These false target sites greatly inhibit GSK3 activity in a competitive manner.[24] This way receptor-bound axin will abolish mediating the phosphorylation of β-catenin. Since beta-catenin is no longer marked for destruction, but continues to be produced, its concentration will increase. Once β-catenin levels rise high enough to saturate all binding sites in the cytoplasm, it will also translocate into the nucleus. Upon engaging the transcription factors LEF1, TCF1, TCF2 or TCF3, β-catenin forces them to disengage their previous partners: Groucho proteins. Unlike Groucho, that recruit transcriptional repressors (e.g. histone-lysine methyltransferases), beta-catenin will bind transcriptional activators, switching on target genes.
3.4. Role in Cell–Cell Adhesion
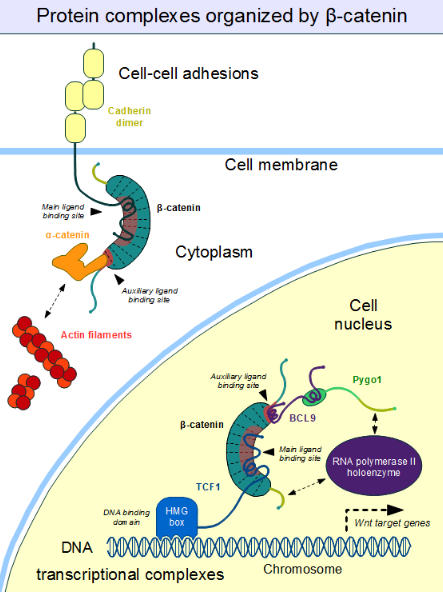
Cell–cell adhesion complexes are essential for the formation of complex animal tissues. β-catenin is part of a protein complex that form adherens junctions.[25] These cell–cell adhesion complexes are necessary for the creation and maintenance of epithelial cell layers and barriers. As a component of the complex, β-catenin can regulate cell growth and adhesion between cells. It may also be responsible for transmitting the contact inhibition signal that causes cells to stop dividing once the epithelial sheet is complete.[26] The E-cadherin – β-catenin – α-catenin complex is weakly associated to actin filaments. Adherens junctions require significant protein dynamics in order to link to the actin cytoskeleton,[25] thereby enabling mechanotransduction.[27]
An important component of the adherens junctions are the cadherin proteins. Cadherins form the cell–cell junctional structures known as adherens junctions as well as the desmosomes. Cadherins are capable of homophilic interactions through their extracellular cadherin repeat domains, in a Ca2+-dependent manner; this can hold adjacent epithelial cells together. While in the adherens junction, cadherins recruit β-catenin molecules onto their intracellular regions[clarification needed]. β-catenin, in turn, associates with another highly dynamic protein, α-catenin, which directly binds to the actin filaments.[28] This is possible because α-catenin and cadherins bind at distinct sites to β-catenin.[29] The β-catenin – α-catenin complex can thus physically form a bridge between cadherins and the actin cytoskeleton.[30] Organization of the cadherin–catenin complex is additionally regulated through phosphorylation and endocytosis of its components.
3.5. Roles in Development
Beta-catenin has a central role in directing several developmental processes, as it can directly bind transcription factors and be regulated by a diffusible extracellular substance: Wnt. It acts upon early embryos to induce entire body regions, as well as individual cells in later stages of development. It also regulates physiological regeneration processes.
Early embryonic patterning
Wnt signaling and beta-catenin dependent gene expression plays a critical role during the formation of different body regions in the early embryo. Experimentally modified embryos that do not express this protein will fail to develop mesoderm and initiate gastrulation.[31] During the blastula and gastrula stages, Wnt as well as BMP and FGF pathways will induce the antero-posterior axis formation, regulate the precise placement of the primitive streak (gastrulation and mesoderm formation) as well as the process of neurulation (central nervous system development).[32]
In Xenopus oocytes, β-catenin is initially equally localized to all regions of the egg, but it is targeted for ubiquitination and degradation by the β-catenin destruction complex. Fertilization of the egg causes a rotation of the outer cortical layers, moving clusters of the Frizzled and Dsh proteins closer to the equatorial region. β-catenin will be enriched locally under the influence of Wnt signaling pathway in the cells that inherit this portion of the cytoplasm. It will eventually translocate to the nucleus to bind TCF3 in order to activate several genes that induce dorsal cell characteristics.[33] This signaling results in a region of cells known as the grey crescent, which is a classical organizer of embryonic development. If this region is surgically removed from the embryo, gastrulation does not occur at all. β-Catenin also plays a crucial role in the induction of the blastopore lip, which in turn initiates gastrulation.[34] Inhibition of GSK-3 translation by injection of antisense mRNA may cause a second blastopore and a superfluous body axis to form. A similar effect can result from the overexpression of β-catenin.[35]
Asymmetric cell division
Beta-catenin has also been implicated in regulation of cell fates through asymmetric cell division in the model organism C. elegans. Similarly to the Xenopus oocytes, this is essentially the result of non-equal distribution of Dsh, Frizzled, axin and APC in the cytoplasm of the mother cell.[36]
Stem cell renewal
One of the most important results of Wnt signaling and the elevated level of beta-catenin in certain cell types is the maintenance of pluripotency.[32] In other cell types and developmental stages, β-catenin may promote differentiation, especially towards mesodermal cell lineages.
Epithelial-to-mesenchymal transition
Beta-catenin also acts as a morphogen in later stages of embryonic development. Together with TGF-β, an important role of β-catenin is to induce a morphogenic change in epithelial cells. It induces them to abandon their tight adhesion and assume a more mobile and loosely associated mesenchymal phenotype. During this process, epithelial cells lose expression of proteins like E-cadherin, Zonula occludens 1 (ZO1), and cytokeratin. At the same time they turn on the expression of vimentin, alpha smooth muscle actin (ACTA2), and fibroblast-specific protein 1 (FSP1). They also produce extracellular matrix components, such as type I collagen and fibronectin. Aberrant activation of the Wnt pathway has been implicated in pathological processes such as fibrosis and cancer.[37] In cardiac muscle development, beta-catenin performs a biphasic role. Initially, the activation of Wnt/beta-catenin is essential for committing mesenchymal cells to a cardiac lineage; however, in later stages of development, the downregulation of beta-catenin is required.[31][38][39]
Involvement in cardiac physiology
In cardiac muscle, beta-catenin forms a complex with N-cadherin at adherens junctions within intercalated disc structures, which are responsible for electrical and mechanical coupling of adjacent cardiac cells. Studies in a model of adult rat ventricular cardiomyocytes have shown that the appearance and distribution of beta-catenin is spatio-temporally regulated during the redifferentiation of these cells in culture. Specifically, beta-catenin is part of a distinct complex with N-cadherin and alpha-catenin, which is abundant at adherens junctions in early stages following cardiomyocyte isolation for the reformation of cell–cell contacts.[40] It has been shown that beta-catenin forms a complex with emerin in cardiomyocytes at adherens junctions within intercalated discs; and this interaction is dependent on the presence of GSK 3-beta phosphorylation sites on beta-catenin. Knocking out emerin significantly altered beta-catenin localization and the overall intercalated disc architecture, which resembled a dilated cardiomyopathy phenotype.[41]
In animal models of cardiac disease, functions of beta-catenin have been unveiled. In a guinea pig model of aortic stenosis and left ventricular hypertrophy, beta-catenin was shown to change subcellular localization from intercalated discs to the cytosol, despite no change in the overall cellular abundance of beta-catenin. vinculin showed a similar profile of change. N-cadherin showed no change, and there was no compensatory upregulation of plakoglobin at intercalated discs in the absence of beta-catenin.[42] In a hamster model of cardiomyopathy and heart failure, cell–cell adhesions were irregular and disorganized, and expression levels of adherens junction/intercalated disc and nuclear pools of beta-catenin were decreased.[43] These data suggest that a loss of beta-catenin may play a role in the diseased intercalated discs that have been associated with cardiac muscle hypertrophy and heart failure. In a rat model of myocardial infarction, adenoviral gene transfer of nonphosphorylatable, constitutively-active beta-catenin decreased MI size, activated the cell cycle, and reduced the amount of apoptosis in cardiomyocytes and cardiac myofibroblasts. This finding was coordinate with enhanced expression of pro-survival proteins, survivin and Bcl-2, and vascular endothelial growth factor while promoting the differentiation of cardiac fibroblasts into myofibroblasts. These findings suggest that beta-catenin can promote the regeneration and healing process following myocardial infarction.[44] In a spontaneously-hypertensive heart failure rat model, investigators detected a shuttling of beta-catenin from the intercalated disc/sarcolemma to the nucleus, evidenced by a reduction of beta-catenin expression in the membrane protein fraction and an increase in the nuclear fraction. Additionally, they found a weakening in the association between glycogen synthase kinase-3β and beta-catenin, which may indicate altered protein stability. Overall, results suggest that an enhanced nuclear localization of beta-catenin may be important in the progression of cardiac hypertrophy.[45]
Regarding the mechanistic role of beta-catenin in cardiac hypertrophy, transgenic mouse studies have shown somewhat conflicting results regarding whether upregulation of beta-catenin is beneficial or detrimental.[46][47][48] A recent study using a conditional knockout mouse that either lacked beta-catenin altogether or expressed a non-degradable form of beta-catenin in cardiomyocytes reconciled a potential reason for these discrepancies. There appears to be strict control over the subcellular localization of beta-catenin in cardiac muscle. Mice lacking beta-catenin had no overt phenotype in the left ventricular myocardium; however, mice harboring a stabilized form of beta-catenin developed dilated cardiomyopathy, suggesting that the temporal regulation of beta-catenin by protein degradation mechanisms is critical for normal functioning of beta-catenin in cardiac cells.[49] In a mouse model harboring knockout of a desmosomal protein, plakoglobin, implicated in arrhythmogenic right ventricular cardiomyopathy, the stabilization of beta-catenin was also enhanced, presumably to compensate for the loss of its plakogloblin homolog. These changes were coordinate with Akt activation and glycogen synthase kinase 3β inhibition, suggesting once again that the abnormal stabilization of beta-catenin may be involved in the development of cardiomyopathy.[50] Further studies employing a double knockout of plakoglobin and beta-catenin showed that the double knockout developed cardiomyopathy, fibrosis and arrhythmias resulting in sudden cardiac death. Intercalated disc architecture was severely impaired and connexin 43-resident gap junctions were markedly reduced. Electrocardiogram measurements captured spontaneous lethal ventricular arrhythmias in the double transgenic animals, suggesting that the two catenins—beta-catenin and plakoglobin are critical and idispensible for mechanoelectrical coupling in cardiomyocytes.[51]
4. Clinical Significance
4.1. Role in Depression
Whether or not a given individual's brain can deal effectively with stress, and thus their susceptibility to depression, depends on the beta-catenin in each person's brain, according to a study conducted at the Icahn School of Medicine at Mount Sinai and published November 12, 2014 in the journal Nature.[52] Higher beta-catenin signaling increases behavioral flexibility, whereas defective beta-catenin signaling leads to depression and reduced stress management.[52]
4.2. Role in Cardiac Disease
Altered expression profiles in beta-catenin have been associated with dilated cardiomyopathy in humans. Beta-catenin upregulation of expression has generally been observed in patients with dilated cardiomyopathy.[53] In a particular study, patients with end-stage dilated cardiomyopathy showed almost doubled estrogen receptor alpha (ER-alpha) mRNA and protein levels, and the ER-alpha/beta-catenin interaction, present at intercalated discs of control, non-diseased human hearts was lost, suggesting that the loss of this interaction at the intercalated disc may play a role in the progression of heart failure.[54] Together with BCL9 and PYGO proteins, beta-catenin coordinates different aspects of heard development, and mutations in Bcl9 or Pygo in model organisms - such as the mouse and zebrafish - cause phenotypes that are very similar to human congenital heart disorders.[55]
4.3. Involvement in Cancer
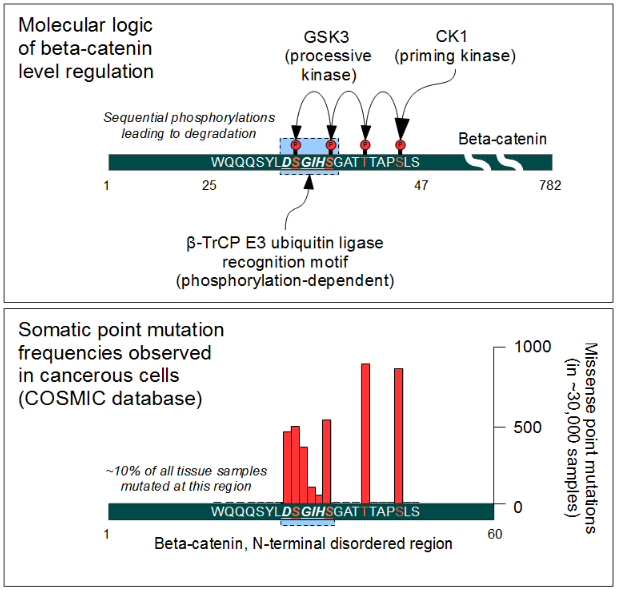
Beta-catenin is a proto-oncogene. Mutations of this gene are commonly found in a variety of cancers: in primary hepatocellular carcinoma, colorectal cancer, ovarian carcinoma, breast cancer, lung cancer and glioblastoma. It has been estimated that approximately 10% of all tissue samples sequenced from all cancers display mutations in the CTNNB1 gene.[56] Most of these mutations cluster on a tiny area of the N-terminal segment of β-catenin: the β-TrCP binding motif. Loss-of-function mutations of this motif essentially make ubiquitinylation and degradation of β-catenin impossible. It will cause β-catenin to translocate to the nucleus without any external stimulus and continuously drive transcription of its target genes. Increased nuclear β-catenin levels have also been noted in basal cell carcinoma (BCC),[57] head and neck squamous cell carcinoma (HNSCC), prostate cancer (CaP),[58] pilomatrixoma (PTR)[59] and medulloblastoma (MDB)[60] These observations may or may not implicate a mutation in the β-catenin gene: other Wnt pathway components can also be faulty.
Similar mutations are also frequently seen in the β-catenin recruiting motifs of APC. Hereditary loss-of-function mutations of APC cause a condition known as Familial Adenomatous Polyposis. Affected individuals develop hundreds of polyps in their large intestine. Most of these polyps are benign in nature, but they have the potential to transform into deadly cancer as time progresses. Somatic mutations of APC in colorectal cancer are also not uncommon.[61] Beta-catenin and APC are among the key genes (together with others, like K-Ras and SMAD4) involved in colorectal cancer development. The potential of β-catenin to change the previously epithelial phenotype of affected cells into an invasive, mesenchyme-like type contributes greatly to metastasis formation.
4.4. As a Therapeutic Target
Due to its involvement in cancer development, inhibition of beta-catenin continues to receive significant attention. But the targeting of the binding site on its armadillo domain is not the simplest task, due to its extensive and relatively flat surface. However, for an efficient inhibition, binding to smaller "hotspots" of this surface is sufficient. This way, a "stapled" helical peptide derived from the natural β-catenin binding motif found in LEF1 was sufficient for the complete inhibition of β-catenin dependent transcription. Recently, several small-molecule compounds have also been developed to target the same, highly positively charged area of the ARM domain (CGP049090, PKF118-310, PKF115-584 and ZTM000990). In addition, β-catenin levels can also be influenced by targeting upstream components of the Wnt pathway as well as the β-catenin destruction complex.[62] The additional N-terminal binding pocket is also important for Wnt target gene activation (required for BCL9 recruitment). This site of the ARM domain can be pharmacologically targeted by carnosic acid, for example.[63] That "auxiliary" site is another attractive target for drug development.[64] Despite intensive preclinical research, no β-catenin inhibitors are available as therapeutic agents yet. However, its function can be further examined by siRNA knockdown based on an independent validation.[65] Another therapeutic approach for reducing β-catenin nuclear accumulation is via the inhibition of galectin-3.[66] The galectin-3 inhibitor GR-MD-02 is currently undergoing clinical trials in combination with the FDA-approved dose of ipilimumab in patients who have advanced melanoma.[67] The proteins BCL9 and BCL9L have been proposed as therapeutic targets for colorectal cancers which present hyper-activated Wnt signaling, because their deletion does not perturb normal homeostasis but strongly affects metastases behaviour.[68]
4.5. Role in Fetal Alcohol Syndrome
β-catenin destabilization by ethanol is one of two known pathways whereby alcohol exposure induces fetal alcohol syndrome (the other is ethanol-induced folate deficiency). Ethanol leads to β-catenin destabilization via a G-protein-dependent pathway, wherein activated Phospholipase Cβ hydrolyzes phosphatidylinositol-(4,5)-bisphosphate to diacylglycerol and inositol-(1,4,5)-trisphosphate. Soluble inositol-(1,4,5)-trisphosphate triggers calcium to be released from the endoplasmic reticulum. This sudden increase in cytoplasmic calcium activates Ca2+/calmodulin-dependent protein kinase (CaMKII). Activated CaMKII destabilizes β-catenin via a poorly characterized mechanism, but which likely involves β-catenin phosphorylation by CaMKII. The β-catenin transcriptional program (which is required for normal neural crest cell development) is thereby suppressed, resulting in premature neural crest cell apoptosis (cell death).[69]
5. Interactions
Beta-catenin has been shown to interact with:
- APC,[70][71][72][73][74][75][76][77]
- AXIN1,[78][79]
- Androgen receptor,[80][81][82][83][84][85]
- CBY1,[86]
- CDH1,[14][71][87][88][89][90][91][92][93][94][95][96][97][98][99][100][101][102][103][104][105][106][107]
- CDH2,[40][108][109]
- CDH3,[106][110]
- CDK5R1,[111]
- CHUK,[112]
- CTNND1,[71][92]
- CTNNA1,[88][97][113][114][115]
- EGFR,[92][101][116]
- Emerin[117][118]
- ESR1[54]
- FHL2,[119]
- GSK3B,[73][120]
- HER2/neu,[93][116][121]
- HNF4A,[84]
- IKK2,[112]
- LEF1,[122][123][124][125]
- MAGI1,[102]
- MUC1,[94][126][127][128][129][130][131]
- NR5A1,[132][133]
- PCAF,[134]
- PHF17,[135]
- Plakoglobin,[71][92]
- PTPN14,[136]
- PTPRF,[93][137]
- PTPRK (PTPkappa),[138]
- PTPRT (PTPrho),[139]
- PTPRU (PCP-2),[140][141][142]
- PSEN1,[143][144][145]
- PTK7[146]
- RuvB-like 1,[147]
- SMAD7,[122]
- SMARCA4[148]
- SLC9A3R1,[96]
- USP9X,[149] and
- VE-cadherin.[150][151]
- XIRP1[152]
References
- "A homolog of the armadillo protein in Drosophila (plakoglobin) associated with E-cadherin". Science 254 (5036): 1359–61. November 1991. doi:10.1126/science.1962194. PMID 1962194. Bibcode: 1991Sci...254.1359M. https://dx.doi.org/10.1126%2Fscience.1962194
- "From cadherins to catenins: cytoplasmic protein interactions and regulation of cell adhesion". Trends in Genetics 9 (9): 317–21. September 1993. doi:10.1016/0168-9525(93)90250-l. PMID 8236461. https://dx.doi.org/10.1016%2F0168-9525%2893%2990250-l
- "Terminal regions of beta-catenin come into view". Structure 16 (3): 336–8. March 2008. doi:10.1016/j.str.2008.02.005. PMID 18334207. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2329800
- "Crystal structure of a full-length beta-catenin". Structure 16 (3): 478–87. March 2008. doi:10.1016/j.str.2007.12.021. PMID 18334222. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4267759
- "Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity". Journal of Biological Chemistry 282 (15): 11221–9. April 2007. doi:10.1074/jbc.M611871200. PMID 17287208. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1850976
- "The WNT/β-catenin dependent transcription: A tissue-specific business". Wiley Interdisciplinary Reviews. Systems Biology and Medicine 13 (3): e1511. October 2020. doi:10.1002/wsbm.1511. PMID 33085215. https://dx.doi.org/10.1002%2Fwsbm.1511
- "The C-terminal transactivation domain of beta-catenin is necessary and sufficient for signaling by the LEF-1/beta-catenin complex in Xenopus laevis". Mechanisms of Development 81 (1–2): 65–74. March 1999. doi:10.1016/s0925-4773(98)00225-1. PMID 10330485. https://dx.doi.org/10.1016%2Fs0925-4773%2898%2900225-1
- "Differential interaction of plakoglobin and beta-catenin with the ubiquitin-proteasome system". Oncogene 19 (16): 1992–2001. April 2000. doi:10.1038/sj.onc.1203519. PMID 10803460. https://dx.doi.org/10.1038%2Fsj.onc.1203519
- "Plakoglobin: role in tumorigenesis and metastasis". International Journal of Cell Biology 2012: 1–14. 2012. doi:10.1155/2012/189521. PMID 22481945. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3312339
- "Mechanistic insights from structural studies of beta-catenin and its binding partners". Journal of Cell Science 120 (Pt 19): 3337–44. October 2007. doi:10.1242/jcs.013771. PMID 17881495. https://dx.doi.org/10.1242%2Fjcs.013771
- "Crystal structure of a beta-catenin/Tcf complex". Cell 103 (6): 885–96. December 2000. doi:10.1016/S0092-8674(00)00192-6. PMID 11136974. https://dx.doi.org/10.1016%2FS0092-8674%2800%2900192-6
- "Tcf4 can specifically recognize beta-catenin using alternative conformations". Nature Structural Biology 8 (12): 1048–52. December 2001. doi:10.1038/nsb718. PMID 11713475. https://dx.doi.org/10.1038%2Fnsb718
- "Structure of a human Tcf4-beta-catenin complex". Nature Structural Biology 8 (12): 1053–7. December 2001. doi:10.1038/nsb720. PMID 11713476. https://dx.doi.org/10.1038%2Fnsb720
- "The structure of the beta-catenin/E-cadherin complex and the molecular basis of diverse ligand recognition by beta-catenin". Cell 105 (3): 391–402. May 2001. doi:10.1016/S0092-8674(01)00330-0. PMID 11348595. https://dx.doi.org/10.1016%2FS0092-8674%2801%2900330-0
- "Crystal structure of a beta-catenin/axin complex suggests a mechanism for the beta-catenin destruction complex". Genes & Development 17 (22): 2753–64. November 2003. doi:10.1101/gad.1142603. PMID 14600025. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=280624
- "Messing up disorder: how do missense mutations in the tumor suppressor protein APC lead to cancer?". Molecular Cancer 10 (1): 101. August 2011. doi:10.1186/1476-4598-10-101. PMID 21859464. PMC 3170638. http://www.molecular-cancer.com/content/pdf/1476-4598-10-101.pdf.
- "Wnt/wingless signaling requires BCL9/legless-mediated recruitment of pygopus to the nuclear beta-catenin-TCF complex". Cell 109 (1): 47–60. April 2002. doi:10.1016/S0092-8674(02)00679-7. PMID 11955446. https://www.zora.uzh.ch/id/eprint/966/1/Krawnt02v.pdf.
- "Structure of the dimerization and beta-catenin-binding region of alpha-catenin". Molecular Cell 5 (3): 533–43. March 2000. doi:10.1016/S1097-2765(00)80447-5. PMID 10882138. https://dx.doi.org/10.1016%2FS1097-2765%2800%2980447-5
- "Crystal structure of a beta-catenin/BCL9/Tcf4 complex". Molecular Cell 24 (2): 293–300. October 2006. doi:10.1016/j.molcel.2006.09.001. PMID 17052462. https://dx.doi.org/10.1016%2Fj.molcel.2006.09.001
- "Essential role of BCL9-2 in the switch between beta-catenin's adhesive and transcriptional functions". Genes & Development 18 (18): 2225–30. September 2004. doi:10.1101/gad.317604. PMID 15371335. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=517514
- "The third 20 amino acid repeat is the tightest binding site of APC for beta-catenin". Journal of Molecular Biology 360 (1): 133–44. June 2006. doi:10.1016/j.jmb.2006.04.064. PMID 16753179. https://dx.doi.org/10.1016%2Fj.jmb.2006.04.064
- "beta-catenin destruction complex: insights and questions from a structural perspective". Oncogene 25 (57): 7482–91. December 2006. doi:10.1038/sj.onc.1210055. PMID 17143292. https://dx.doi.org/10.1038%2Fsj.onc.1210055
- "Dishevelled interacts with the DIX domain polymerization interface of Axin to interfere with its function in down-regulating β-catenin". Proceedings of the National Academy of Sciences of the United States of America 108 (5): 1937–42. February 2011. doi:10.1073/pnas.1017063108. PMID 21245303. Bibcode: 2011PNAS..108.1937F. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3033301
- "Inhibition of GSK3 by Wnt signalling--two contrasting models". Journal of Cell Science 124 (Pt 21): 3537–44. November 2011. doi:10.1242/jcs.091991. PMID 22083140. https://dx.doi.org/10.1242%2Fjcs.091991
- "Balancing cell adhesion and Wnt signaling, the key role of beta-catenin". Current Opinion in Genetics & Development 16 (1): 51–9. February 2006. doi:10.1016/j.gde.2005.12.007. PMID 16377174. https://dx.doi.org/10.1016%2Fj.gde.2005.12.007
- "Entrez Gene: catenin (cadherin-associated protein)". https://www.ncbi.nlm.nih.gov/sites/entrez?Db=gene&Cmd=ShowDetailView&TermToSearch=1499.
- "An ensemble of flexible conformations underlies mechanotransduction by the cadherin-catenin adhesion complex". Proc Natl Acad Sci USA 116 (43): 21545–21555. October 22, 2019. doi:10.1073/pnas.1911489116. PMID 31591245. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=6815173
- "Activated nanoscale actin-binding domain motion in the catenin-cadherin complex revealed by neutron spin echo spectroscopy". Proc. Natl. Acad. Sci. U.S.A. 118 (13): e2025012118. March 30, 2021. doi:10.1073/pnas.2025012118. PMID 33753508. Bibcode: 2021PNAS..11825012F. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=8020631
- "Regulation of cell–cell adhesion by the cadherin-catenin complex". Biochemical Society Transactions 36 (Pt 2): 149–55. April 2008. doi:10.1042/BST0360149. PMID 18363555. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3368607
- "beta-Catenin: a pivot between cell adhesion and Wnt signalling". Current Biology 15 (2): R64–7. January 2005. doi:10.1016/j.cub.2004.12.058. PMID 15668160. https://dx.doi.org/10.1016%2Fj.cub.2004.12.058
- "Lack of beta-catenin affects mouse development at gastrulation". Development 121 (11): 3529–37. November 1995. doi:10.1242/dev.121.11.3529. PMID 8582267. https://dx.doi.org/10.1242%2Fdev.121.11.3529
- "Maintaining embryonic stem cell pluripotency with Wnt signaling". Development 138 (20): 4341–50. October 2011. doi:10.1242/dev.066209. PMID 21903672. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3177306
- "Beta-catenin translocation into nuclei demarcates the dorsalizing centers in frog and fish embryos". Mechanisms of Development 57 (2): 191–8. July 1996. doi:10.1016/0925-4773(96)00546-1. PMID 8843396. https://dx.doi.org/10.1016%2F0925-4773%2896%2900546-1
- "Establishment of the dorso-ventral axis in Xenopus embryos is presaged by early asymmetries in beta-catenin that are modulated by the Wnt signaling pathway". The Journal of Cell Biology 136 (5): 1123–36. March 1997. doi:10.1083/jcb.136.5.1123. PMID 9060476. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2132470
- "Induction of a secondary embryonic axis in zebrafish occurs following the overexpression of beta-catenin". Mechanisms of Development 53 (2): 261–73. October 1995. doi:10.1016/0925-4773(95)00442-4. PMID 8562427. https://dx.doi.org/10.1016%2F0925-4773%2895%2900442-4
- "Control of cell polarity and asymmetric division in C. elegans". Planar Cell Polarity During Development. 101. 2012. 55–76. doi:10.1016/B978-0-12-394592-1.00003-X. ISBN 9780123945921. https://dx.doi.org/10.1016%2FB978-0-12-394592-1.00003-X
- "E-cadherin/β-catenin complex and the epithelial barrier". Journal of Biomedicine & Biotechnology 2011: 1–6. 2011. doi:10.1155/2011/567305. PMID 22007144. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3191826
- "Role of beta-catenin in adult cardiac remodeling". Cell Cycle 6 (17): 2120–6. September 2007. doi:10.4161/cc.6.17.4632. PMID 17786052. https://dx.doi.org/10.4161%2Fcc.6.17.4632
- "Formation of multiple hearts in mice following deletion of beta-catenin in the embryonic endoderm". Developmental Cell 3 (2): 171–81. August 2002. doi:10.1016/s1534-5807(02)00206-x. PMID 12194849. https://dx.doi.org/10.1016%2Fs1534-5807%2802%2900206-x
- "N-cadherin in adult rat cardiomyocytes in culture. II. Spatio-temporal appearance of proteins involved in cell–cell contact and communication. Formation of two distinct N-cadherin/catenin complexes". Journal of Cell Science 109 ( Pt 1) (1): 11–20. January 1996. doi:10.1242/jcs.109.1.11. PMID 8834786. https://dx.doi.org/10.1242%2Fjcs.109.1.11
- "Identification of an emerin-beta-catenin complex in the heart important for intercalated disc architecture and beta-catenin localisation". Cellular and Molecular Life Sciences 67 (5): 781–96. March 2010. doi:10.1007/s00018-009-0219-8. PMID 19997769. https://dx.doi.org/10.1007%2Fs00018-009-0219-8
- "Chronic pressure overload cardiac hypertrophy and failure in guinea pigs: III. Intercalated disc remodeling". Journal of Molecular and Cellular Cardiology 31 (2): 333–43. February 1999. doi:10.1006/jmcc.1998.0886. PMID 10093046. https://dx.doi.org/10.1006%2Fjmcc.1998.0886
- "Alterations in adhesion junction precede gap junction remodelling during the development of heart failure in cardiomyopathic hamsters". Cardiovascular Research 92 (1): 95–105. October 2011. doi:10.1093/cvr/cvr182. PMID 21693625. https://dx.doi.org/10.1093%2Fcvr%2Fcvr182
- "Beta-catenin overexpression reduces myocardial infarct size through differential effects on cardiomyocytes and cardiac fibroblasts". The Journal of Biological Chemistry 281 (41): 30979–89. October 2006. doi:10.1074/jbc.M603916200. PMID 16920707. https://dx.doi.org/10.1074%2Fjbc.M603916200
- "Expression and redistribution of β-catenin in the cardiac myocytes of left ventricle of spontaneously hypertensive rat". Journal of Molecular Histology 44 (5): 565–73. October 2013. doi:10.1007/s10735-013-9507-6. PMID 23591738. https://dx.doi.org/10.1007%2Fs10735-013-9507-6
- "Beta-catenin downregulation is required for adaptive cardiac remodeling". Circulation Research 100 (9): 1353–62. May 2007. doi:10.1161/01.RES.0000266605.63681.5a. PMID 17413044. https://dx.doi.org/10.1161%2F01.RES.0000266605.63681.5a
- "The beta-catenin/T-cell factor/lymphocyte enhancer factor signaling pathway is required for normal and stress-induced cardiac hypertrophy". Molecular and Cellular Biology 26 (12): 4462–73. June 2006. doi:10.1128/MCB.02157-05. PMID 16738313. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1489123
- "Stabilization of beta-catenin by a Wnt-independent mechanism regulates cardiomyocyte growth". Proceedings of the National Academy of Sciences of the United States of America 100 (8): 4610–5. April 2003. doi:10.1073/pnas.0835895100. PMID 12668767. Bibcode: 2003PNAS..100.4610H. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=153603
- "Stabilised beta-catenin in postnatal ventricular myocardium leads to dilated cardiomyopathy and premature death". Basic Research in Cardiology 105 (5): 597–608. September 2010. doi:10.1007/s00395-010-0101-8. PMID 20376467. http://www.zora.uzh.ch/id/eprint/34179/1/Hirschy.pdf.
- "Cardiac tissue-restricted deletion of plakoglobin results in progressive cardiomyopathy and activation of -catenin signaling". Molecular and Cellular Biology 31 (6): 1134–44. March 2011. doi:10.1128/MCB.01025-10. PMID 21245375. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3067899
- "Loss of cadherin-binding proteins β-catenin and plakoglobin in the heart leads to gap junction remodeling and arrhythmogenesis". Molecular and Cellular Biology 32 (6): 1056–67. March 2012. doi:10.1128/MCB.06188-11. PMID 22252313. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3295003
- "β-catenin mediates stress resilience through Dicer1/microRNA regulation". Nature 516 (7529): 51–5. December 2014. doi:10.1038/nature13976. PMID 25383518. Bibcode: 2014Natur.516...51D. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4257892
- "Dilated cardiomyopathy: a disease of the intercalated disc?". Trends in Cardiovascular Medicine 13 (1): 30–8. January 2003. doi:10.1016/s1050-1738(02)00209-8. PMID 12554098. https://dx.doi.org/10.1016%2Fs1050-1738%2802%2900209-8
- "Estrogen receptor alpha up-regulation and redistribution in human heart failure". FASEB Journal 20 (7): 926–34. May 2006. doi:10.1096/fj.05-5148com. PMID 16675850. https://dx.doi.org/10.1096%2Ffj.05-5148com
- "Pygo genes cause congenital heart defects by tissue-specific perturbation of Wnt/β-catenin signaling". Genes & Development 32 (21–22): 1443–1458. November 2018. doi:10.1101/gad.315531.118. PMID 30366904. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=6217730
- "COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer". Nucleic Acids Research 39 (Database issue): D945–50. January 2011. doi:10.1093/nar/gkq929. PMID 20952405. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3013785
- "Nuclear beta-catenin in basal cell carcinoma correlates with increased proliferation". The British Journal of Dermatology 151 (1): 157–64. July 2004. doi:10.1111/j.1365-2133.2004.06048.x. PMID 15270885. https://dx.doi.org/10.1111%2Fj.1365-2133.2004.06048.x
- "Wnt/β-catenin signalling in prostate cancer". Nature Reviews. Urology 9 (8): 418–28. August 2012. doi:10.1038/nrurol.2012.116. PMID 22710668. https://dx.doi.org/10.1038%2Fnrurol.2012.116
- "beta-Catenin is expressed aberrantly in tumors expressing shadow cells. Pilomatricoma, craniopharyngioma, and calcifying odontogenic cyst". American Journal of Clinical Pathology 120 (5): 732–6. November 2003. doi:10.1309/EALEG7LD6W7167PX. PMID 14608900. https://dx.doi.org/10.1309%2FEALEG7LD6W7167PX
- "beta-Catenin status predicts a favorable outcome in childhood medulloblastoma: the United Kingdom Children's Cancer Study Group Brain Tumour Committee". Journal of Clinical Oncology 23 (31): 7951–7. November 2005. doi:10.1200/JCO.2005.01.5479. PMID 16258095. https://dx.doi.org/10.1200%2FJCO.2005.01.5479
- "Nuclear translocation of beta-catenin in colorectal cancer". British Journal of Cancer 82 (10): 1689–93. May 2000. doi:10.1054/bjoc.1999.1112. PMID 10817505. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2374509
- "Wnt/beta-catenin signaling and small molecule inhibitors". Current Pharmaceutical Design 19 (4): 634–64. 2013. doi:10.2174/1381612811306040634. PMID 23016862. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3529405
- "An intrinsically labile α-helix abutting the BCL9-binding site of β-catenin is required for its inhibition by carnosic acid". Nature Communications 3 (2): 680. February 2012. doi:10.1038/ncomms1680. PMID 22353711. Bibcode: 2012NatCo...3..680D. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3293410
- "Targeted disruption of the BCL9/β-catenin complex inhibits oncogenic Wnt signaling". Science Translational Medicine 4 (148): 148ra117. August 2012. doi:10.1126/scitranslmed.3003808. PMID 22914623. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3631420
- "Validation of RNAi Silencing Efficiency Using Gene Array Data shows 18.5% Failure Rate across 429 Independent Experiments" (in en). Molecular Therapy. Nucleic Acids 5 (9): e366. September 2016. doi:10.1038/mtna.2016.66. PMID 27673562. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=5056990
- "Endogenous and exogenous galectin-3 promote the adhesion of tumor cells with low expression of MUC1 to HUVECs through upregulation of N-cadherin and CD44". Laboratory Investigation 98 (12): 1642–1656. 2018-08-31. doi:10.1038/s41374-018-0119-3. PMID 30171204. https://dx.doi.org/10.1038%2Fs41374-018-0119-3
- Clinical trial number NCT02117362 for "Galectin Inhibitor (GR-MD-02) and Ipilimumab in Patients With Metastatic Melanoma" at ClinicalTrials.gov https://www.clinicaltrials.gov/show/NCT02117362
- "BCL9/9L-β-catenin Signaling is Associated With Poor Outcome in Colorectal Cancer". EBioMedicine 2 (12): 1932–43. December 2015. doi:10.1016/j.ebiom.2015.10.030. PMID 26844272. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4703711
- "Calcium-mediated repression of β-catenin and its transcriptional signaling mediates neural crest cell death in an avian model of fetal alcohol syndrome". Birth Defects Research. Part A, Clinical and Molecular Teratology 91 (7): 591–602. July 2011. doi:10.1002/bdra.20833. PMID 21630427. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4827605
- "Association of the APC tumor suppressor protein with catenins". Science 262 (5140): 1734–7. December 1993. doi:10.1126/science.8259519. PMID 8259519. Bibcode: 1993Sci...262.1734S. https://dx.doi.org/10.1126%2Fscience.8259519
- "Expression and interaction of different catenins in colorectal carcinoma cells". International Journal of Molecular Medicine 8 (6): 695–8. December 2001. doi:10.3892/ijmm.8.6.695. PMID 11712088. https://dx.doi.org/10.3892%2Fijmm.8.6.695
- "Differences between the interaction of beta-catenin with non-phosphorylated and single-mimicked phosphorylated 20-amino acid residue repeats of the APC protein". Journal of Molecular Biology 327 (2): 359–67. March 2003. doi:10.1016/S0022-2836(03)00144-X. PMID 12628243. https://dx.doi.org/10.1016%2FS0022-2836%2803%2900144-X
- "The interaction between beta-catenin, GSK3beta and APC after motogen induced cell–cell dissociation, and their involvement in signal transduction pathways in prostate cancer". International Journal of Oncology 18 (4): 843–7. April 2001. doi:10.3892/ijo.18.4.843. PMID 11251183. https://dx.doi.org/10.3892%2Fijo.18.4.843
- "Pin1 regulates turnover and subcellular localization of beta-catenin by inhibiting its interaction with APC". Nature Cell Biology 3 (9): 793–801. September 2001. doi:10.1038/ncb0901-793. PMID 11533658. https://dx.doi.org/10.1038%2Fncb0901-793
- "Association and regulation of casein kinase 2 activity by adenomatous polyposis coli protein". Proceedings of the National Academy of Sciences of the United States of America 99 (9): 5959–64. April 2002. doi:10.1073/pnas.092143199. PMID 11972058. Bibcode: 2002PNAS...99.5959K. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=122884
- "DAP-1, a novel protein that interacts with the guanylate kinase-like domains of hDLG and PSD-95". Genes to Cells 2 (6): 415–24. June 1997. doi:10.1046/j.1365-2443.1997.1310329.x. PMID 9286858. https://dx.doi.org/10.1046%2Fj.1365-2443.1997.1310329.x
- "Molecular mechanisms of beta-catenin recognition by adenomatous polyposis coli revealed by the structure of an APC-beta-catenin complex". The EMBO Journal 20 (22): 6203–12. November 2001. doi:10.1093/emboj/20.22.6203. PMID 11707392. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=125720
- "Axin, an inhibitor of the Wnt signalling pathway, interacts with beta-catenin, GSK-3beta and APC and reduces the beta-catenin level". Genes to Cells 3 (6): 395–403. June 1998. doi:10.1046/j.1365-2443.1998.00198.x. PMID 9734785. https://dx.doi.org/10.1046%2Fj.1365-2443.1998.00198.x
- "Regulation of the Wnt signaling pathway by disabled-2 (Dab2)". The EMBO Journal 22 (12): 3084–94. June 2003. doi:10.1093/emboj/cdg286. PMID 12805222. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=162138
- "Linking beta-catenin to androgen-signaling pathway". The Journal of Biological Chemistry 277 (13): 11336–44. March 2002. doi:10.1074/jbc.M111962200. PMID 11792709. https://dx.doi.org/10.1074%2Fjbc.M111962200
- "Recruitment of beta-catenin by wild-type or mutant androgen receptors correlates with ligand-stimulated growth of prostate cancer cells". Molecular Endocrinology 18 (10): 2388–401. October 2004. doi:10.1210/me.2003-0436. PMID 15256534. https://dx.doi.org/10.1210%2Fme.2003-0436
- "Antiandrogen effects of mifepristone on coactivator and corepressor interactions with the androgen receptor". Molecular Endocrinology 18 (1): 70–85. January 2004. doi:10.1210/me.2003-0189. PMID 14593076. https://dx.doi.org/10.1210%2Fme.2003-0189
- "A direct beta-catenin-independent interaction between androgen receptor and T cell factor 4". The Journal of Biological Chemistry 278 (33): 30828–34. August 2003. doi:10.1074/jbc.M301208200. PMID 12799378. https://dx.doi.org/10.1074%2Fjbc.M301208200
- "Functional localization and competition between the androgen receptor and T-cell factor for nuclear beta-catenin: a means for inhibition of the Tcf signaling axis". Oncogene 22 (36): 5602–13. August 2003. doi:10.1038/sj.onc.1206802. PMID 12944908. https://dx.doi.org/10.1038%2Fsj.onc.1206802
- "Liganded androgen receptor interaction with beta-catenin: nuclear co-localization and modulation of transcriptional activity in neuronal cells". The Journal of Biological Chemistry 277 (23): 20702–10. June 2002. doi:10.1074/jbc.M200545200. PMID 11916967. https://dx.doi.org/10.1074%2Fjbc.M200545200
- "Chibby, a nuclear beta-catenin-associated antagonist of the Wnt/Wingless pathway". Nature 422 (6934): 905–9. April 2003. doi:10.1038/nature01570. PMID 12712206. Bibcode: 2003Natur.422..905T. https://dx.doi.org/10.1038%2Fnature01570
- "HGF/SF modifies the interaction between its receptor c-Met, and the E-cadherin/catenin complex in prostate cancer cells". International Journal of Molecular Medicine 7 (4): 385–8. April 2001. doi:10.3892/ijmm.7.4.385. PMID 11254878. https://dx.doi.org/10.3892%2Fijmm.7.4.385
- "A truncated beta-catenin disrupts the interaction between E-cadherin and alpha-catenin: a cause of loss of intercellular adhesiveness in human cancer cell lines". Cancer Research 54 (23): 6282–7. December 1994. PMID 7954478. http://www.ncbi.nlm.nih.gov/pubmed/7954478
- "Vinculin is associated with the E-cadherin adhesion complex". The Journal of Biological Chemistry 272 (51): 32448–53. December 1997. doi:10.1074/jbc.272.51.32448. PMID 9405455. https://dx.doi.org/10.1074%2Fjbc.272.51.32448
- "Tyrosine phosphorylation regulates the adhesions of ras-transformed breast epithelia". The Journal of Cell Biology 130 (2): 461–71. July 1995. doi:10.1083/jcb.130.2.461. PMID 7542250. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2199929
- "CAS/CSE 1 stimulates E-cadhrin-dependent cell polarity in HT-29 human colon epithelial cells". Biochemical and Biophysical Research Communications 294 (4): 900–5. June 2002. doi:10.1016/S0006-291X(02)00551-X. PMID 12061792. https://dx.doi.org/10.1016%2FS0006-291X%2802%2900551-X
- "The epidermal growth factor receptor modulates the interaction of E-cadherin with the actin cytoskeleton". The Journal of Biological Chemistry 273 (15): 9078–84. April 1998. doi:10.1074/jbc.273.15.9078. PMID 9535896. https://dx.doi.org/10.1074%2Fjbc.273.15.9078
- "Geldanamycin abrogates ErbB2 association with proteasome-resistant beta-catenin in melanoma cells, increases beta-catenin-E-cadherin association, and decreases beta-catenin-sensitive transcription". Cancer Research 61 (4): 1671–7. February 2001. PMID 11245482. http://www.ncbi.nlm.nih.gov/pubmed/11245482
- "Interaction of glycogen synthase kinase 3beta with the DF3/MUC1 carcinoma-associated antigen and beta-catenin". Molecular and Cellular Biology 18 (12): 7216–24. December 1998. doi:10.1128/mcb.18.12.7216. PMID 9819408. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=109303
- "Ksp-cadherin is a functional cell–cell adhesion molecule related to LI-cadherin". Experimental Cell Research 294 (2): 345–55. April 2004. doi:10.1016/j.yexcr.2003.11.022. PMID 15023525. https://dx.doi.org/10.1016%2Fj.yexcr.2003.11.022
- "EBP50, a beta-catenin-associating protein, enhances Wnt signaling and is over-expressed in hepatocellular carcinoma". Hepatology 38 (1): 178–86. July 2003. doi:10.1053/jhep.2003.50270. PMID 12830000. https://dx.doi.org/10.1053%2Fjhep.2003.50270
- "p120 Catenin-associated Fer and Fyn tyrosine kinases regulate beta-catenin Tyr-142 phosphorylation and beta-catenin-alpha-catenin Interaction". Molecular and Cellular Biology 23 (7): 2287–97. April 2003. doi:10.1128/MCB.23.7.2287-2297.2003. PMID 12640114. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=150740
- "Promyogenic members of the Ig and cadherin families associate to positively regulate differentiation". Proceedings of the National Academy of Sciences of the United States of America 100 (7): 3989–94. April 2003. doi:10.1073/pnas.0736565100. PMID 12634428. Bibcode: 2003PNAS..100.3989K. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=153035
- "UCS15A, a novel small molecule, SH3 domain-mediated protein-protein interaction blocking drug". Oncogene 21 (13): 2037–50. March 2002. doi:10.1038/sj.onc.1205271. PMID 11960376. https://dx.doi.org/10.1038%2Fsj.onc.1205271
- "Expression of E- or P-cadherin is not sufficient to modify the morphology and the tumorigenic behavior of murine spindle carcinoma cells. Possible involvement of plakoglobin". Journal of Cell Science 105 ( Pt 4) (4): 923–34. August 1993. doi:10.1242/jcs.105.4.923. PMID 8227214. https://dx.doi.org/10.1242%2Fjcs.105.4.923
- "Induction of tyrosine phosphorylation and association of beta-catenin with EGF receptor upon tryptic digestion of quiescent cells at confluence". Oncogene 15 (1): 71–8. July 1997. doi:10.1038/sj.onc.1201160. PMID 9233779. https://dx.doi.org/10.1038%2Fsj.onc.1201160
- "MAGI-1 interacts with beta-catenin and is associated with cell–cell adhesion structures". Biochemical and Biophysical Research Communications 270 (3): 903–9. April 2000. doi:10.1006/bbrc.2000.2471. PMID 10772923. https://dx.doi.org/10.1006%2Fbbrc.2000.2471
- "Modification of the composition of polycystin-1 multiprotein complexes by calcium and tyrosine phosphorylation". Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 1535 (1): 21–35. December 2000. doi:10.1016/S0925-4439(00)00079-X. PMID 11113628. https://dx.doi.org/10.1016%2FS0925-4439%2800%2900079-X
- "Association of p120, a tyrosine kinase substrate, with E-cadherin/catenin complexes". The Journal of Cell Biology 128 (5): 949–57. March 1995. doi:10.1083/jcb.128.5.949. PMID 7876318. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2120395
- "Tyrosine phosphorylation and dissociation of occludin-ZO-1 and E-cadherin-beta-catenin complexes from the cytoskeleton by oxidative stress". The Biochemical Journal 368 (Pt 2): 471–81. December 2002. doi:10.1042/BJ20011804. PMID 12169098. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1222996
- "The fate of E- and P-cadherin during the early stages of apoptosis". Cell Death and Differentiation 6 (4): 377–86. April 1999. doi:10.1038/sj.cdd.4400504. PMID 10381631. https://dx.doi.org/10.1038%2Fsj.cdd.4400504
- "Inhibition of fibroblast growth factor 19 reduces tumor growth by modulating beta-catenin signaling". Cancer Research 68 (13): 5086–95. July 2008. doi:10.1158/0008-5472.CAN-07-2325. PMID 18593907. https://dx.doi.org/10.1158%2F0008-5472.CAN-07-2325
- "A novel cell–cell junction system: the cortex adhaerens mosaic of lens fiber cells". Journal of Cell Science 116 (Pt 24): 4985–95. December 2003. doi:10.1242/jcs.00815. PMID 14625392. https://dx.doi.org/10.1242%2Fjcs.00815
- "N-cadherin-catenin complexes form prior to cleavage of the proregion and transport to the plasma membrane". The Journal of Biological Chemistry 278 (19): 17269–76. May 2003. doi:10.1074/jbc.M211452200. PMID 12604612. https://dx.doi.org/10.1074%2Fjbc.M211452200
- "Amino-terminal domain of classic cadherins determines the specificity of the adhesive interactions". Journal of Cell Science 113 ( Pt 16) (16): 2829–36. August 2000. doi:10.1242/jcs.113.16.2829. PMID 10910767. https://dx.doi.org/10.1242%2Fjcs.113.16.2829
- "p35/cdk5 binds and phosphorylates beta-catenin and regulates beta-catenin/presenilin-1 interaction". The European Journal of Neuroscience 13 (2): 241–7. January 2001. doi:10.1046/j.1460-9568.2001.01376.x. PMID 11168528. https://dx.doi.org/10.1046%2Fj.1460-9568.2001.01376.x
- "Regulation of beta-catenin function by the IkappaB kinases". The Journal of Biological Chemistry 276 (45): 42276–86. November 2001. doi:10.1074/jbc.M104227200. PMID 11527961. https://dx.doi.org/10.1074%2Fjbc.M104227200
- "A mutation in alpha-catenin disrupts adhesion in clone A cells without perturbing its actin and beta-catenin binding activity". Cell Adhesion and Communication 5 (4): 283–96. June 1998. doi:10.3109/15419069809040298. PMID 9762469. https://dx.doi.org/10.3109%2F15419069809040298
- "Assembly of the cadherin-catenin complex in vitro with recombinant proteins". Journal of Cell Science 107 ( Pt 12) (12): 3655–63. December 1994. doi:10.1242/jcs.107.12.3655. PMID 7706414. https://dx.doi.org/10.1242%2Fjcs.107.12.3655
- "E-cadherin mediated cell adhesion recruits SAP97 into the cortical cytoskeleton". Journal of Cell Science 111 ( Pt 8) (8): 1071–80. April 1998. doi:10.1242/jcs.111.8.1071. PMID 9512503. https://dx.doi.org/10.1242%2Fjcs.111.8.1071
- "ErbB-beta-catenin complexes are associated with human infiltrating ductal breast and murine mammary tumor virus (MMTV)-Wnt-1 and MMTV-c-Neu transgenic carcinomas". The Journal of Biological Chemistry 277 (25): 22692–8. June 2002. doi:10.1074/jbc.M201975200. PMID 11950845. https://dx.doi.org/10.1074%2Fjbc.M201975200
- "Heart-specific localization of emerin: new insights into Emery-Dreifuss muscular dystrophy". Human Molecular Genetics 6 (13): 2257–64. December 1997. doi:10.1093/hmg/6.13.2257. PMID 9361031. https://dx.doi.org/10.1093%2Fhmg%2F6.13.2257
- "The inner nuclear membrane protein emerin regulates beta-catenin activity by restricting its accumulation in the nucleus". The EMBO Journal 25 (14): 3275–85. July 2006. doi:10.1038/sj.emboj.7601230. PMID 16858403. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1523183
- "Identification of the LIM protein FHL2 as a coactivator of beta-catenin". The Journal of Biological Chemistry 278 (7): 5188–94. February 2003. doi:10.1074/jbc.M207216200. PMID 12466281. https://dx.doi.org/10.1074%2Fjbc.M207216200
- "DIX domains of Dvl and axin are necessary for protein interactions and their ability to regulate beta-catenin stability". Molecular and Cellular Biology 19 (6): 4414–22. June 1999. doi:10.1128/mcb.19.6.4414. PMID 10330181. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=104400
- "c-erbB-2 gene product directly associates with beta-catenin and plakoglobin". Biochemical and Biophysical Research Communications 208 (3): 1067–72. March 1995. doi:10.1006/bbrc.1995.1443. PMID 7702605. https://dx.doi.org/10.1006%2Fbbrc.1995.1443
- "Interaction between Smad7 and beta-catenin: importance for transforming growth factor beta-induced apoptosis". Molecular and Cellular Biology 25 (4): 1475–88. February 2005. doi:10.1128/MCB.25.4.1475-1488.2005. PMID 15684397. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=548008
- "A functional screen in human cells identifies UBF2 as an RNA polymerase II transcription factor that enhances the beta-catenin signaling pathway". Molecular and Cellular Biology 23 (11): 3936–50. June 2003. doi:10.1128/MCB.23.11.3936-3950.2003. PMID 12748295. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=155208
- "Functional interaction of beta-catenin with the transcription factor LEF-1". Nature 382 (6592): 638–42. August 1996. doi:10.1038/382638a0. PMID 8757136. Bibcode: 1996Natur.382..638B. https://dx.doi.org/10.1038%2F382638a0
- "Association of Smads with lymphoid enhancer binding factor 1/T cell-specific factor mediates cooperative signaling by the transforming growth factor-beta and wnt pathways". Proceedings of the National Academy of Sciences of the United States of America 97 (15): 8358–63. July 2000. doi:10.1073/pnas.150152697. PMID 10890911. Bibcode: 2000PNAS...97.8358L. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=26952
- "Interaction of the DF3/MUC1 breast carcinoma-associated antigen and beta-catenin in cell adhesion". The Journal of Biological Chemistry 272 (19): 12492–4. May 1997. doi:10.1074/jbc.272.19.12492. PMID 9139698. https://dx.doi.org/10.1074%2Fjbc.272.19.12492
- "Interleukin-7 induces MUC1". Cancer Biology & Therapy 2 (2): 194–5. 2003. doi:10.4161/cbt.2.2.351. PMID 12750562. https://dx.doi.org/10.4161%2Fcbt.2.2.351
- "MUC1 alters beta-catenin-dependent tumor formation and promotes cellular invasion". Oncogene 22 (9): 1324–32. March 2003. doi:10.1038/sj.onc.1206291. PMID 12618757. https://dx.doi.org/10.1038%2Fsj.onc.1206291
- "The c-Src tyrosine kinase regulates signaling of the human DF3/MUC1 carcinoma-associated antigen with GSK3 beta and beta-catenin". The Journal of Biological Chemistry 276 (9): 6061–4. March 2001. doi:10.1074/jbc.C000754200. PMID 11152665. https://dx.doi.org/10.1074%2Fjbc.C000754200
- "Protein kinase C delta regulates function of the DF3/MUC1 carcinoma antigen in beta-catenin signaling". The Journal of Biological Chemistry 277 (20): 17616–22. May 2002. doi:10.1074/jbc.M200436200. PMID 11877440. https://dx.doi.org/10.1074%2Fjbc.M200436200
- "The epidermal growth factor receptor regulates interaction of the human DF3/MUC1 carcinoma antigen with c-Src and beta-catenin". The Journal of Biological Chemistry 276 (38): 35239–42. September 2001. doi:10.1074/jbc.C100359200. PMID 11483589. https://dx.doi.org/10.1074%2Fjbc.C100359200
- "T-cell factor 4N (TCF-4N), a novel isoform of mouse TCF-4, synergizes with beta-catenin to coactivate C/EBPalpha and steroidogenic factor 1 transcription factors". Molecular and Cellular Biology 23 (15): 5366–75. August 2003. doi:10.1128/MCB.23.15.5366-5375.2003. PMID 12861022. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=165725
- "Dax-1 (dosage-sensitive sex reversal-adrenal hypoplasia congenita critical region on the X chromosome, gene 1) gene transcription is regulated by wnt4 in the female developing gonad". Molecular Endocrinology 17 (4): 507–19. April 2003. doi:10.1210/me.2002-0362. PMID 12554773. https://dx.doi.org/10.1210%2Fme.2002-0362
- "PCAF acetylates -catenin and improves its stability". Molecular Biology of the Cell 20 (1): 419–27. January 2009. doi:10.1091/mbc.E08-08-0792. PMID 18987336. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2613091
- "One hit, two outcomes for VHL-mediated tumorigenesis". Nature Cell Biology 10 (10): 1127–8. October 2008. doi:10.1038/ncb1008-1127. PMID 18830218. https://dx.doi.org/10.1038%2Fncb1008-1127
- "The protein tyrosine phosphatase Pez is a major phosphatase of adherens junctions and dephosphorylates beta-catenin". Molecular Biology of the Cell 14 (6): 2520–9. June 2003. doi:10.1091/mbc.E02-09-0577. PMID 12808048. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=194899
- "Cellular redistribution of protein tyrosine phosphatases LAR and PTPsigma by inducible proteolytic processing". The Journal of Cell Biology 138 (3): 681–96. August 1997. doi:10.1083/jcb.138.3.681. PMID 9245795. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2141638
- "Association of human protein-tyrosine phosphatase kappa with members of the armadillo family". The Journal of Biological Chemistry 271 (28): 16712–9. July 1996. doi:10.1074/jbc.271.28.16712. PMID 8663237. https://dx.doi.org/10.1074%2Fjbc.271.28.16712
- "Intracellular substrates of brain-enriched receptor protein tyrosine phosphatase rho (RPTPrho/PTPRT)". Brain Research 1116 (1): 50–7. October 2006. doi:10.1016/j.brainres.2006.07.122. PMID 16973135. https://dx.doi.org/10.1016%2Fj.brainres.2006.07.122
- "Molecular cloning and characterization of a novel human receptor protein tyrosine phosphatase gene, hPTP-J: down-regulation of gene expression by PMA and calcium ionophore in Jurkat T lymphoma cells". Biochemical and Biophysical Research Communications 231 (1): 77–81. February 1997. doi:10.1006/bbrc.1997.6004. PMID 9070223. https://dx.doi.org/10.1006%2Fbbrc.1997.6004
- "Physical and functional interaction between receptor-like protein tyrosine phosphatase PCP-2 and beta-catenin". Biochemistry 41 (52): 15854–60. December 2002. doi:10.1021/bi026095u. PMID 12501215. https://dx.doi.org/10.1021%2Fbi026095u
- "Structural basis of interaction between protein tyrosine phosphatase PCP-2 and beta-catenin". Science in China Series C: Life Sciences 48 (2): 163–7. April 2005. doi:10.1007/bf02879669. PMID 15986889. https://dx.doi.org/10.1007%2Fbf02879669
- "Abrogation of the presenilin 1/beta-catenin interaction and preservation of the heterodimeric presenilin 1 complex following caspase activation". The Journal of Biological Chemistry 273 (51): 33909–14. December 1998. doi:10.1074/jbc.273.51.33909. PMID 9852041. https://dx.doi.org/10.1074%2Fjbc.273.51.33909
- "Presenilin 1 facilitates the constitutive turnover of beta-catenin: differential activity of Alzheimer's disease-linked PS1 mutants in the beta-catenin-signaling pathway". The Journal of Neuroscience 19 (11): 4229–37. June 1999. doi:10.1523/JNEUROSCI.19-11-04229.1999. PMID 10341227. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=6782616
- "Direct association of presenilin-1 with beta-catenin". FEBS Letters 433 (1–2): 73–7. August 1998. doi:10.1016/S0014-5793(98)00886-2. PMID 9738936. https://dx.doi.org/10.1016%2FS0014-5793%2898%2900886-2
- "Protein tyrosine kinase 7 has a conserved role in Wnt/β-catenin canonical signalling". EMBO Reports 12 (1): 43–9. January 2011. doi:10.1038/embor.2010.185. PMID 21132015. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3024124
- "Pontin52, an interaction partner of beta-catenin, binds to the TATA box binding protein". Proceedings of the National Academy of Sciences of the United States of America 95 (25): 14787–92. December 1998. doi:10.1073/pnas.95.25.14787. PMID 9843967. Bibcode: 1998PNAS...9514787B. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=24527
- "The chromatin remodelling factor Brg-1 interacts with beta-catenin to promote target gene activation". The EMBO Journal 20 (17): 4935–43. September 2001. doi:10.1093/emboj/20.17.4935. PMID 11532957. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=125268
- "The deubiquitinating enzyme Fam interacts with and stabilizes beta-catenin". Genes to Cells 4 (12): 757–67. December 1999. doi:10.1046/j.1365-2443.1999.00297.x. PMID 10620020. https://dx.doi.org/10.1046%2Fj.1365-2443.1999.00297.x
- "Alteration of interendothelial adherens junctions following tumor cell–endothelial cell interaction in vitro". Experimental Cell Research 237 (2): 347–56. December 1997. doi:10.1006/excr.1997.3799. PMID 9434630. https://dx.doi.org/10.1006%2Fexcr.1997.3799
- "Histamine stimulates phosphorylation of adherens junction proteins and alters their link to vimentin". American Journal of Physiology. Lung Cellular and Molecular Physiology 282 (6): L1330–8. June 2002. doi:10.1152/ajplung.00329.2001. PMID 12003790. https://dx.doi.org/10.1152%2Fajplung.00329.2001
- "Localization of the novel Xin protein to the adherens junction complex in cardiac and skeletal muscle during development". Developmental Dynamics 225 (1): 1–13. September 2002. doi:10.1002/dvdy.10131. PMID 12203715. https://dx.doi.org/10.1002%2Fdvdy.10131

