 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sirius Huang | -- | 1743 | 2022-11-30 01:40:33 |
Video Upload Options
Forkhead box protein O1 (FOXO1) also known as forkhead in rhabdomyosarcoma (FKHR) is a protein that in humans is encoded by the FOXO1 gene. FOXO1 is a transcription factor that plays important roles in regulation of gluconeogenesis and glycogenolysis by insulin signaling, and is also central to the decision for a preadipocyte to commit to adipogenesis. It is primarily regulated through phosphorylation on multiple residues; its transcriptional activity is dependent on its phosphorylation state.
1. Function
1.1. Adipogenesis
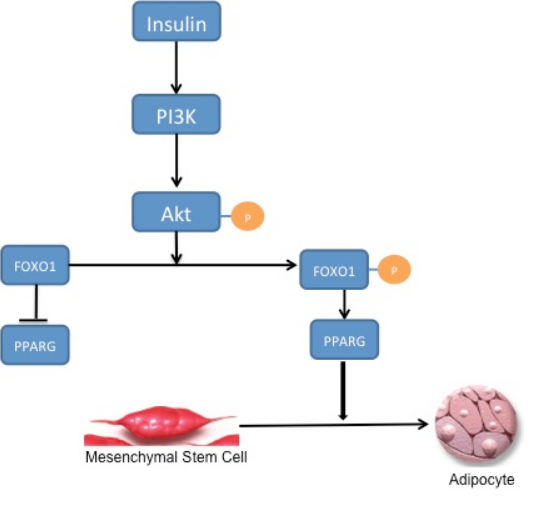
FOXO1 negatively regulates adipogenesis.[1] Presently, the exact mechanism by which this is accomplished is not entirely understood. In the currently accepted model, FOXO1 negatively regulates adipogenesis by binding to the promoter sites of PPARG and preventing its transcription. Rising levels of PPARG are required to initiate adipogenesis; by preventing its transcription, FOXO1 is preventing the onset of adipogenesis. During stimulation by insulin, FOXO1 is excluded from the nucleus and is subsequently unable to prevent transcription of PPARG and inhibit adipogenesis.[2] However, there is substantial evidence to suggest that there are other factors that mediate the interaction between FOXO1 and the PPARG promoter, and that inhibition of adipogenesis is not entirely dependent on FOXO1 preventing transcription of PPARG.[3] The failure to commit to adipogenesis is primarily due to active FOXO1 arresting the cell in G0/G1 through activation of yet unknown downstream targets, with a putative target being SOD2.[4]
FOXO1 belongs to the forkhead family of transcription factors that are characterized by a distinct fork head domain. The specific function of this gene has not yet been determined; however, it may play a role in myogenic growth and differentiation.[5] FOXO1 is essential for the maintenance of human ESC pluripotency. This function is probably mediated through direct control by FOXO1 of OCT4 and SOX2 gene expression through occupation and activation of their respective promoters.[6] In hepatic cells this transcription factor seems to increase the expression of PEPCK and glycogen-6-phosphatase (the same enzymes that are blocked via the metformin/AMPK/SHP pathway). Blocking this transcription factor offers an opportunity for novel therapies for diabetes mellitus.[7] In pancreatic alpha-cells FOXO1 is important in regulating prepro-glucagon expression.[8] In pancreatic beta cells FOXO1 mediates glucagon-like peptide-1 effects on pancreatic beta-cell mass.[9]
1.2. Gluconeogenesis and Glycogenolysis
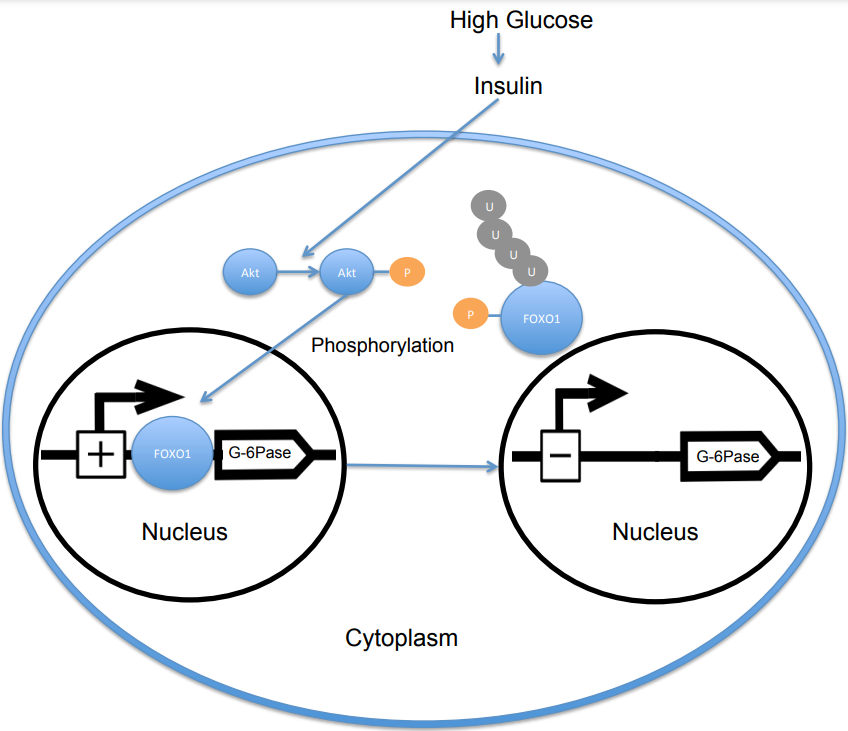
When the level of blood glucose is high, the pancreas releases insulin into the bloodstream. Insulin then causes the activation of PI3K, which subsequently phosphorylates Akt. Akt then phosphorylates FOXO1, causing nuclear exclusion.[10][11] This phosphorylated FOXO1 is then ubiquitinated and degraded by the proteosome.[12] The phosphorylation of FOXO1 is irreversible; this prolongs insulin's inhibitory effect on glucose metabolism and hepatic glucose production. Transcription of glucose 6-phosphatase subsequently decreases, which consequently decreases the rates of gluconeogenesis and glycogenolysis.[13] FOXO1 also activates transcription of phosphoenolpyruvate carboxykinase, which is required for gluconeogenesis.[14] The activity of FOXO1 is also regulated through CBP induced acetylation[15] on Lys-242, Lys-245, and Lys-262. These lysine residues are located within the DNA-binding domain; acetylation inhibits the ability of FOXO1 to interact with the glucose-6 phosphatase promoter by decreasing the stability of the FOXO1-DNA complex. Additionally, this acetylation increases the rate of phosphorylation on Ser-253 by Akt. Mutating Ser-253 to Ala-253 makes FOXO1 constitutively active. SIRT1 reverses this acetylation process; however, the exact mechanism by which SIRT1 deacetylates FOXO1 is still under investigation; presently, acetylation is thought to mitigate the transcriptional activity of FOXO1 and thereby provide an additional level of metabolic regulation that is independent of the insulin/PI3K pathway.[16]
1.3. Apoptosis
FOXO1 may play an important role in apoptosis because it is phosphorylated and inhibited by AKT.[17] When FOXO1 over expressed in human LNCaP prostate cancer cells, it caused apoptosis in these cancer cells.[17] Also, It is detected that FOXO1 regulateTNF-related apoptosis-inducing ligand (TRAIL), which cause FOXO1-induced apoptosis in the human prostate cancer cell line LAPC4 when FOXO1 adenovirus-mediated overexpression was used.[17] FOXO1 upregulate Fas ligand (FasL) transcriptionally that result in promotes apoptotic cell death.[17] Additionally, FOXO1 trans-activate Bim protein, which a member of the Bcl-2 family that promotes apoptosis and plays a role in the intrinsic mitochondrial apoptotic pathway.[17] Further, it was revealed that DNA damage-induced cell death in p53-deficient and p53-proficient cells reduced when human FOXO1 silenced by siRNA.[17]
In type 2 diabetes the beta cells of the pancreas, which normally produce insulin undergo apoptosis, which greatly reduces the production of insulin. Fatty acids in the beta cells activate FOXO1, resulting in apopotosis of the beta cells.[18]
1.4. Cell Cycle Regulation
FOXO1 activation plays a role in cell cycle progression regulation.[17] The transcription and half- life of cyclin-dependent kinase inhibitor p27KIP1 rises when FOXO1 is active.[17] A study detects that FOXO1 regulates the nuclear localization of p27KIP1 in porcine granulosa cells and impacts cell cycle progression.[17] Furthermore, FOXO1-mediated cell cycle arrest is linked with cyclin D1 and cyclin D2 suppression in mammals.[17] It was detected that human FOXO1 is linked with the cyclin D1 promoter using chromatin immunoprecipitation assays (ChIP assays).[17] H215R is a human FOXO1 mutant, which cannot bind to the canonical FRE to induce expression of p27KIP1, repress cyclin D1 and cyclin D2 promoter activity and encourages cell cycle arrest at cyclin G1 (CCNG1).[17] As a result of that, activation of FOXO1 prevents the cell-division cycle at cyclin G1 (CCNG1) out of one of two ways stimulating or suppressing gene transcription.[17]
2. Mechanism of Action
In its un-phosphorylated state, FOXO1 is localized to the nucleus, where it binds to the insulin response sequence located in the promoter for glucose 6-phosphatase and increases its rate of transcription. FOXO1, through increasing transcription of glucose-6-phosphatase, indirectly increases the rate of hepatic glucose production.[14] However, when FOXO1 is phosphorylated by Akt on Thr-24, Ser-256, and Ser-319, it is excluded from the nucleus, where it is then ubiquitinated and degraded. The phosphorylation of FOXO1 by Akt subsequently decreases the hepatic glucose production through a decrease in transcription of glucose 6-phosphatase.
3. Regulation
There are three processes, namely acetylation, phosphorylation, and ubiquitination that are responsible for regulation of the activity of forkhead box O1 (FOXO1).[19]
3.1. Phosphorylation
Phosphorylation of the FOXO1 protein is a result of the activation of the PI3K /AKT pathway.[19] Serum and glucocorticoid-inducible kinase SGK can also phosphorylate and inactivate FOXO1 transcription factor.[17] FOXO1 translocate from the nucleus to cytoplasm and inactivate through phosphorylation at well-defined sites by AKT/SGK1 protein kinases.[19] FOXO1 transcription factor can phosphorylate directly by AKT/SGK1 on three sites T24, S256 and S319.[20] Additionally, FOXO1 loses its interactions with DNA when phosphorylated by AKT/SGK1 because S256, which is one of the three AKT/SGK sites, changes the DNA-binding domain charge from a positive charge to a negative charge.[19]
Insulin signaling substrates 1 and 2 of the insulin-signaling cascade also regulate FOXO1 through phosphorylation by AKT.[19] AKT, which is referred to as protein kinase B, phosphorylates FOXO1 and accumulates in the cytosol.[19]
Casein kinase 1, a growth factor-activated protein kinase, also phosphorylates and potentiates FOXO1 and translocates FOXO1 to the cytoplasm.[19]
4. Research
Because FOXO1 provides a link between transcription and metabolic control by insulin, it is also a potential target for genetic control of type 2 diabetes. In the insulin-resistant murine model, there is increased hepatic glucose production due to a loss in insulin sensitivity; the rates of hepatic gluconeogenesis and glycogenolysis are increased when compared to normal mice; this is presumably due to un-regulated FOXO1. When the same experiment was repeated with haploinsufficient FOXO1, insulin sensitivity was partially restored, and hepatic glucose production subsequently decreased.[21] Similarly, in mice fed with a high fat diet (HFD), there is increased insulin resistance in skeletal and liver cells. However, when haploinsufficient FOXO1 mice were treated with the same HFD, there was a notable decrease in insulin resistance in both skeletal and liver cells. This effect was significantly augmented by the simultaneous administration of rosiglitazone, which is a commonly prescribed anti-diabetic drug.[22] These results create an opportunity for a novel gene therapy based approach to alleviating insulin desensitization in type 2 diabetes.
In diabetes (both type 1 and type 2), gluconeogenesis in the kidney contributes more to blood glucose than it does in normal subjects.[23] Enhancing suppression of FOXO1 by insulin can reduce gluconeogenesis in both the liver and kidney.[23]
In HFD-fed mice, the combination of FOXO1 and Notch-1 haploinsufficiency was more effective at restoring insulin sensitivity than FOXO1 haploinsufficiency alone.[24]
Insulin-producing cells could be generated through the inhibition of FOXO1 in intestinal organoids generated from intestinal stem cells isolated from adult tissue.[25]
5. Clinical Significance
- Translocation of this gene with PAX3 has been associated with alveolar rhabdomyosarcoma.[26][27]
- In Gluconeogenesis, FOXO1 gene regulates the glucose levels due to the low output of hepatic glucose.[19] In mice, it cuts fasting blood glucose levels by inhibiting formulation of the gluconeogenic genes.[19]
- FOXO1 plays a role in the protection of cells from oxidative stress.[19] It seems to promote cell death when oxidative stress is high in tissues that are involved in diabetic complications.[19] In such situations, it has a destructive role instead of a protective role.[19]
- FOXO1 helps in wound healing in mice through coordination of response of keratinocytes and functions in keratinocytes to bring down oxidative stress.[19] Wound healing is a very complicated biological process and studies have indicated that FOXO1 transcription factor helps in orchestrating events that enhance the healing process in keratinocytes.[28] Localization of FOXO1 nuclear increased four times in wound-healing keratinocytes.[28] It encourages the migration of the keratinocytes through upregulating the growth factor.[28]
- In the Innate Immune system, FOXO1 has been proved to enhance inflammation through increasing formulation of several proinflammatory genes.[19] It mediates formulation of proinflammatory cytokines in response to high glucose levels, TNF and LPS stimulation.[19]
- In Adaptive Immunity system, FOXO1 regulates the return of peripheral B cells by upregulation of L-section and controls class-switch recombination of peripheral B cells and in T cells it enhances survival of CD8 memory.[19]
- In Carcinogenesis, FOXO1 plays a role of a tumor suppressor and its inactivation has been documented in many kinds of human cancer.[19] It suppresses survival of tumor cells by inducing apoptosis in prostate cancer cells and glioma cells by upregulating the proapoptotic factors.[19] Increased activation of FOXO1 may inhibit the metastasis of the prostate cancer cells to other organs by suppressing the migration and invasion or suppressing the Runt-domain containing Runx2 transcriptional activity.[19]
6. Interactions
FOXO1 has been shown to interact with:
References
- "The forkhead transcription factor Foxo1: a possible link between obesity and insulin resistance". Molecular Cell 11 (1): 6–8. January 2003. doi:10.1016/S1097-2765(03)00003-0. PMID 12535515. https://dx.doi.org/10.1016%2FS1097-2765%2803%2900003-0
- "FOXO1 represses peroxisome proliferator-activated receptor-gamma1 and -gamma2 gene promoters in primary adipocytes. A novel paradigm to increase insulin sensitivity". The Journal of Biological Chemistry 281 (29): 19881–91. July 2006. doi:10.1074/jbc.M600320200. PMID 16670091. https://dx.doi.org/10.1074%2Fjbc.M600320200
- "Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction". Nature 423 (6939): 550–5. May 2003. doi:10.1038/nature01667. PMID 12754525. https://dx.doi.org/10.1038%2Fnature01667
- "The forkhead transcription factor FoxO1 regulates proliferation and transdifferentiation of hepatic stellate cells". Gastroenterology 132 (4): 1434–46. April 2007. doi:10.1053/j.gastro.2007.01.033. PMID 17408630. https://zenodo.org/record/1187188/files/article.pdf.
- "Entrez Gene: FOXO1 forkhead box O1 (rhabdomyosarcoma)". https://www.ncbi.nlm.nih.gov/sites/entrez?Db=gene&Cmd=ShowDetailView&TermToSearch=2308.
- "FOXO1 is an essential regulator of pluripotency in human embryonic stem cells". Nature Cell Biology 13 (9): 1092–9. July 2011. doi:10.1038/ncb2293. PMID 21804543. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4053529
- "Discovery of novel forkhead box O1 inhibitors for treating type 2 diabetes: improvement of fasting glycemia in diabetic db/db mice". Molecular Pharmacology 78 (5): 961–70. November 2010. doi:10.1124/mol.110.065714. PMID 20736318. https://dx.doi.org/10.1124%2Fmol.110.065714
- "FoxO1 is required for the regulation of preproglucagon gene expression by insulin in pancreatic alphaTC1-9 cells". The Journal of Biological Chemistry 281 (51): 39358–69. December 2006. doi:10.1074/jbc.M605022200. PMID 17062568. https://dx.doi.org/10.1074%2Fjbc.M605022200
- "Transcription factor FoxO1 mediates glucagon-like peptide-1 effects on pancreatic beta-cell mass". Diabetes 55 (5): 1190–6. May 2006. doi:10.2337/db05-0825. PMID 16644672. https://dx.doi.org/10.2337%2Fdb05-0825
- "Two novel phosphorylation sites on FKHR that are critical for its nuclear exclusion". The EMBO Journal 21 (9): 2263–71. May 2002. doi:10.1093/emboj/21.9.2263. PMID 11980723. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=125977
- "Roles of the forkhead in rhabdomyosarcoma (FKHR) phosphorylation sites in regulating 14-3-3 binding, transactivation and nuclear targetting". The Biochemical Journal 354 (Pt 3): 605–12. March 2001. doi:10.1042/0264-6021:3540605. PMID 11237865. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1221692
- "Insulin-induced phosphorylation of FKHR (Foxo1) targets to proteasomal degradation". Proceedings of the National Academy of Sciences of the United States of America 100 (20): 11285–90. September 2003. doi:10.1073/pnas.1934283100. PMID 13679577. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=208749
- "FOXO transcription factors in the regulatory networks of longevity". Journal of Biochemistry 141 (6): 769–74. June 2007. doi:10.1093/jb/mvm104. PMID 17569704. https://dx.doi.org/10.1093%2Fjb%2Fmvm104
- "The forkhead transcription factor Foxo1 (Fkhr) confers insulin sensitivity onto glucose-6-phosphatase expression". The Journal of Clinical Investigation 108 (9): 1359–67. November 2001. doi:10.1172/JCI12876. PMID 11696581. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=209440
- "Acetylation of Foxo1 alters its DNA-binding ability and sensitivity to phosphorylation". Proceedings of the National Academy of Sciences of the United States of America 102 (32): 11278–83. August 2005. doi:10.1073/pnas.0502738102. PMID 16076959. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1183558
- "SIRT2 regulates adipocyte differentiation through FoxO1 acetylation/deacetylation". Cell Metabolism 6 (2): 105–14. August 2007. doi:10.1016/j.cmet.2007.07.003. PMID 17681146. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2083635
- "FOXO1: a potential target for human diseases". Current Drug Targets 12 (9): 1235–44. August 2011. doi:10.2174/138945011796150280. PMID 21443466. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4591039
- "Death versus dedifferentiation: The molecular bases of beta cell mass reduction in type 2 diabetes". Seminars in Cell and Developmental Biology. 2019. doi:10.1016/j.semcdb.2019.12.002. PMID 31831356. https://dx.doi.org/10.1016%2Fj.semcdb.2019.12.002
- "FOXO transcription factors: their clinical significance and regulation". BioMed Research International 2014: 925350. April 2014. doi:10.1155/2014/925350. PMID 24864265. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4016844
- "FOXO transcription factors throughout T cell biology". Nature Reviews. Immunology 12 (9): 649–61. September 2012. doi:10.1038/nri3278. PMID 22918467. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3875397
- "Regulation of insulin action and pancreatic beta-cell function by mutated alleles of the gene encoding forkhead transcription factor Foxo1". Nature Genetics 32 (2): 245–53. October 2002. doi:10.1038/ng890. PMID 12219087. https://dx.doi.org/10.1038%2Fng890
- "FoxO1 haploinsufficiency protects against high-fat diet-induced insulin resistance with enhanced peroxisome proliferator-activated receptor gamma activation in adipose tissue". Diabetes 58 (6): 1275–82. June 2009. doi:10.2337/db08-1001. PMID 19289458. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2682681
- "Molecular signaling mechanisms of renal gluconeogenesis in nondiabetic and diabetic conditions". Journal of Cellular Physiology 234 (6): 8134-8151. 2019. doi:10.1002/jcp.27598. PMID 30370538. https://dx.doi.org/10.1002%2Fjcp.27598
- "Inhibition of Notch signaling ameliorates insulin resistance in a FoxO1-dependent manner". Nature Medicine 17 (8): 961–7. July 2011. doi:10.1038/nm.2378. PMID 21804540. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3387563
- "FOXO1 inhibition yields functional insulin-producing cells in human gut organoid cultures". Nature Communications 5: 4242. June 2014. doi:10.1038/ncomms5242. PMID 24979718. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4083475
- "Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma". Nature Genetics 5 (3): 230–5. November 1993. doi:10.1038/ng1193-230. PMID 8275086. https://dx.doi.org/10.1038%2Fng1193-230
- "PAX3-FOXO1 fusion gene in rhabdomyosarcoma". Cancer Letters 270 (1): 10–8. October 2008. doi:10.1016/j.canlet.2008.03.035. PMID 18457914. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2575376
- "Impact of Diabetes on the Protective Role of FOXO1 in Wound Healing". Journal of Dental Research 94 (8): 1025–6. August 2015. doi:10.1177/0022034515586353. PMID 25978971. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=530387
- "AKT-independent protection of prostate cancer cells from apoptosis mediated through complex formation between the androgen receptor and FKHR". Molecular and Cellular Biology 23 (1): 104–18. January 2003. doi:10.1128/MCB.23.1.104-118.2003. PMID 12482965. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=140652
- "Ligand-dependent interaction of estrogen receptor-alpha with members of the forkhead transcription factor family". The Journal of Biological Chemistry 276 (36): 33554–60. September 2001. doi:10.1074/jbc.M105555200. PMID 11435445. https://dx.doi.org/10.1074%2Fjbc.M105555200
- "DAF-16 recruits the CREB-binding protein coactivator complex to the insulin-like growth factor binding protein 1 promoter in HepG2 cells". Proceedings of the National Academy of Sciences of the United States of America 97 (19): 10412–7. September 2000. doi:10.1073/pnas.190326997. PMID 10973497. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=27038
- "Interaction of FoxO1 and TSC2 induces insulin resistance through activation of the mammalian target of rapamycin/p70 S6K pathway". The Journal of Biological Chemistry 281 (52): 40242–51. December 2006. doi:10.1074/jbc.M608116200. PMID 17077083. https://dx.doi.org/10.1074%2Fjbc.M608116200

