Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Dharmendra Kumar Yadav | -- | 2367 | 2022-11-30 04:18:50 | | | |
| 2 | Catherine Yang | Meta information modification | 2367 | 2022-11-30 04:55:22 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Singh, D.D.; Shati, A.A.; Alfaifi, M.Y.; Elbehairi, S.E.I.; Han, I.; Choi, E.; Yadav, D.K. Type-2 Diabetes Mellitus and Dementia. Encyclopedia. Available online: https://encyclopedia.pub/entry/37224 (accessed on 23 July 2026).
Singh DD, Shati AA, Alfaifi MY, Elbehairi SEI, Han I, Choi E, et al. Type-2 Diabetes Mellitus and Dementia. Encyclopedia. Available at: https://encyclopedia.pub/entry/37224. Accessed July 23, 2026.
Singh, Desh Deepak, Ali A. Shati, Mohammad Y. Alfaifi, Serag Eldin I. Elbehairi, Ihn Han, Eun-Ha Choi, Dharmendra K. Yadav. "Type-2 Diabetes Mellitus and Dementia" Encyclopedia, https://encyclopedia.pub/entry/37224 (accessed July 23, 2026).
Singh, D.D., Shati, A.A., Alfaifi, M.Y., Elbehairi, S.E.I., Han, I., Choi, E., & Yadav, D.K. (2022, November 30). Type-2 Diabetes Mellitus and Dementia. In Encyclopedia. https://encyclopedia.pub/entry/37224
Singh, Desh Deepak, et al. "Type-2 Diabetes Mellitus and Dementia." Encyclopedia. Web. 30 November, 2022.
Copy Citation
Dementia is reported to be common in those with type 2 diabetes mellitus. Type 2 diabetes contributes to common molecular mechanisms and an underlying pathology with dementia. Brain cells becoming resistant to insulin leads to elevated blood glucose levels, impaired synaptic plasticity, microglial overactivation, mitochondrial dysfunction, neuronal apoptosis, nutrient deprivation, TAU (Tubulin-Associated Unit) phosphorylation, and cholinergic dysfunction.
type-2 diabetes mellitus
dementia
insulin signaling
1. The Glucose Transporter’s Function in Cognition
T2DM increases the risk of cognitive impairment and IR is linked to a more rapid reduction in the memory-encoding mechanism and thinking skills [1]. Various types of genes, including SLC2A1–SLC2A14 and molecules (GLUT1–GLUT14), are actively involved in the transport of glucose in various regions of the brain and each cell type expresses multiple proteins [2]. GLUT proteins are classified into three classes. Proteins GLUT 1–GLUT 4 and GLUT 14 are classified as group I, GLUT 5, GLUT 7, GLUT 9, and GLUT 11 are classified as group 2, and GLUT 6, GLUT 8, GLUT 10, GLUT 12, and GLUT 13 are classified as group 3 [3][4]. GLUT 2 controls energy regulation, neurotransmitter release, and glucose release in glial cells. Brain stem nuclei and tanycytic, vagus motor nucleus, astrocytes, hypothalamus, arcuate nucleus, olfactory bulbs, nucleus tractus solitarius, paraventricular hypothalamic nucleus, lateral hypothalamic area, and neurons all contain GLUT 2 [4]. GLUT 3 is found in cell bodies, neurons, and dendrites; brain micro vessels; and brain astroglia cells. It has been observed that insulin accelerates the translocation of GLUT 3 and increases glucose uptake by neurons [5]. Glucose entry into the cells is carried out by GLUT 4, which is mainly found in the hippocampal region and maintains insulin regulation and improves cognitive development. It also acts as an insulin-sensitive glucose transporter [6]. There is evidence of GLUT 5 in microglial cells. The brain has a low fructose concentration, and glucose transport activity is substantially lower than that of fructose. Studies on animal models have demonstrated that fructose can pass across the BBB and be used as an energy source by brain cells [1]. The importance of GLUT 5 in the brain remains unclear and requires further investigation. GLUT 6 is involved in the nervous system’s physiological activity and transports hexoses across the membrane [4].
2. Insulin Signaling and Neuro-Complications
IR in dementia is caused by the amyloid precursor protein GSK3 (Glycogen synthase kinase 3) enzyme, involved in glycogen metabolism, oxidative stress, mitochondrial dysfunction, brain inflammation, ion channel activation, and the Shc family of signaling adaptor proteins. IR causes cell breakdown and destruction as well as increased glucose uptake, metabolism, and intake, all of which contribute to abnormal tau aggregation, inhibit lipolysis, and inhibit gluconeogenesis (Figure 1) [7]. If brain cells becoming too resistant to insulin leads to elevated blood glucose, impaired synaptic plasticity, microglial overactivation, mitochondrial dysfunction, neuronal apoptosis, nutrient deprivation, TAU phosphorylation, and cholinergic dysfunction, dementia is a group of symptoms affecting memory-encoding mechanisms, including difficulty in visual and spatial abilities, problem-solving, handling complex tasks, planning and organizing, coordination and motor functions, loss of memory, and changes in cognitive functions. Advanced dementia may develop into Alzheimer’s disease (AD). Dementia patients have synuclein aggregates and plaque accumulation, blood–brain barrier leakage, and neuroinflammation (Figure 2). Disintegrating microtubules and amyloid beta plaques are formed in AD (Figure 3). Sporadic forms of dementia are more common; both semantic and episodic memory are caused by cognitive impairment and visuospatial impairment. Motor coordination is also affected in severe cases of disease [8]. IRs affect intellectual ability, increase the generation of excitability, and promote memory consolidation. IR in cognitive impairment is signified by mitochondrial dysfunction, which is involved in neurodegeneration by reducing glucose transport and inducing the formation of phosphorylated tau protein (Figure 1) [9]. The activation of IR autophosphorylation (IRP) leads to the tyrosine phosphorylation of IRS-1 (insulin receptor substrate 1), which activates PI3K (phosphoinositide 3-kinase) and decreases synaptic plasticity and memory [9][10]. The NMDARs (N-methyl-D-aspartate receptors) are activated by calcium ion channels and activated signaling is linked to the development of neurological complications. NMDAR and IR-dependent signaling molecular mechanisms resulted in amyloid oligomer accumulation, increased TNF-α release, and increased concentrations of stress-induced JNK (Jun N-terminal kinase), resulting in IRS-1 inhibitory phosphorylation. The amyloid β oligomers activate further extracellular exclusion of IRs from the cell surface. All these events block the neuronal regulation of insulin, leading to impaired synaptic plasticity (Figure 4) [9][10]. Dementia is a group of symptoms affecting memory-encoding mechanisms, including difficulty in visual and spatial abilities, problem-solving, handling complex tasks, planning, and organizing, coordination and motor functions, loss of memory, and changes in cognitive functions. Advanced dementia can progress to Alzheimer’s disease [6]. Dementia patients have synuclein aggregates and plaque accumulation, blood–brain barrier leakage, and neuroinflammation, as shown in Figure 5. Disintegrating microtubules and amyloid beta plaques are formed in AD, as shown in Figure 2.
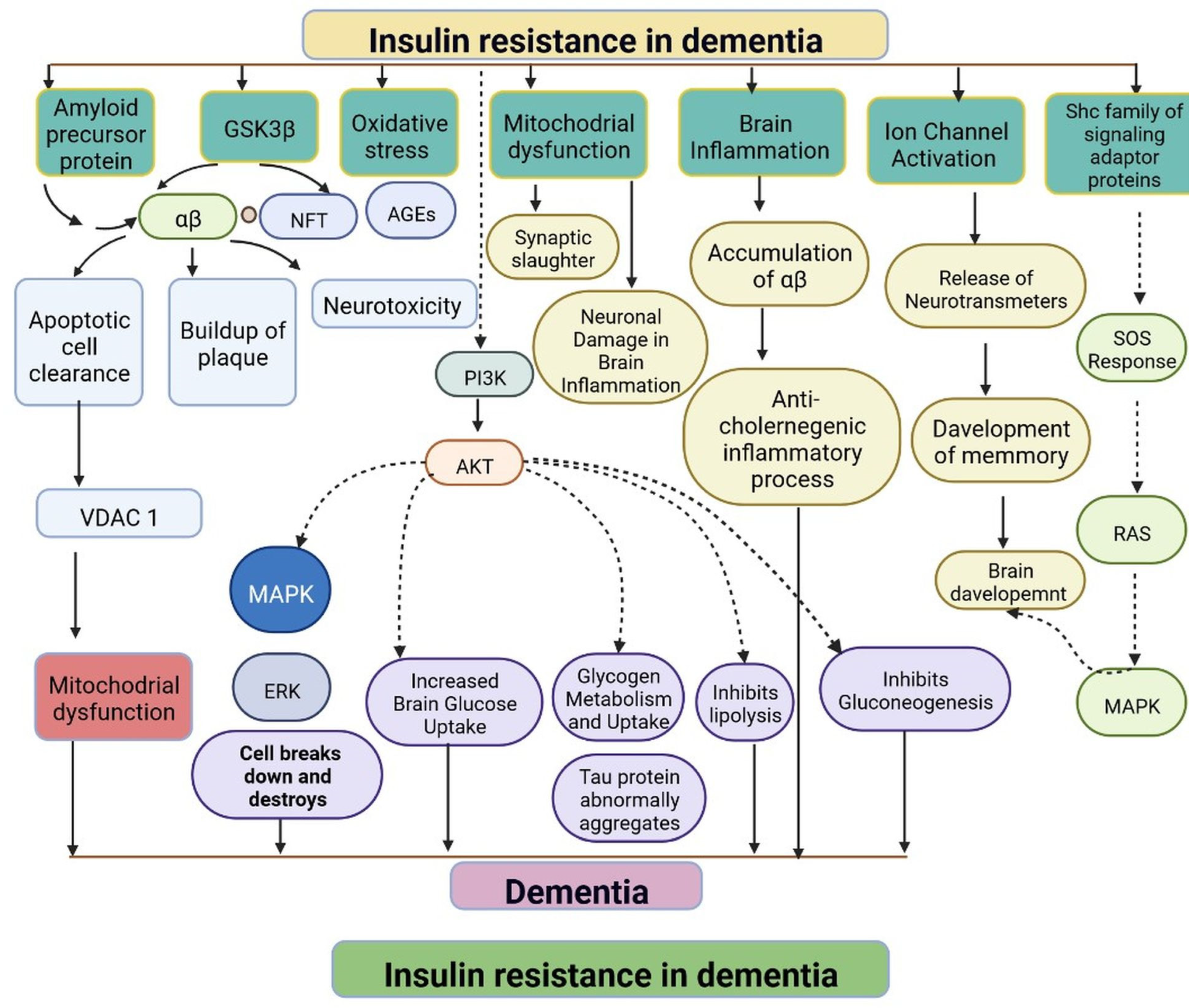
Figure 1. Representation of IR in dementia: resistance to insulin leads to mitochondrial dysfunction, damage to brain cells, altered brain glucose, aggregation of tau protein, inhibition of the lipolysis process, and an anticholinergic inflammatory process. The dotted lines represent inhibitory mechanisms.

Figure 2. The pathological properties of the Alzheimer’s disease brain (involving tau protein) compared with that of the healthy brain, on various magnification levels. It can be adapted as a component.
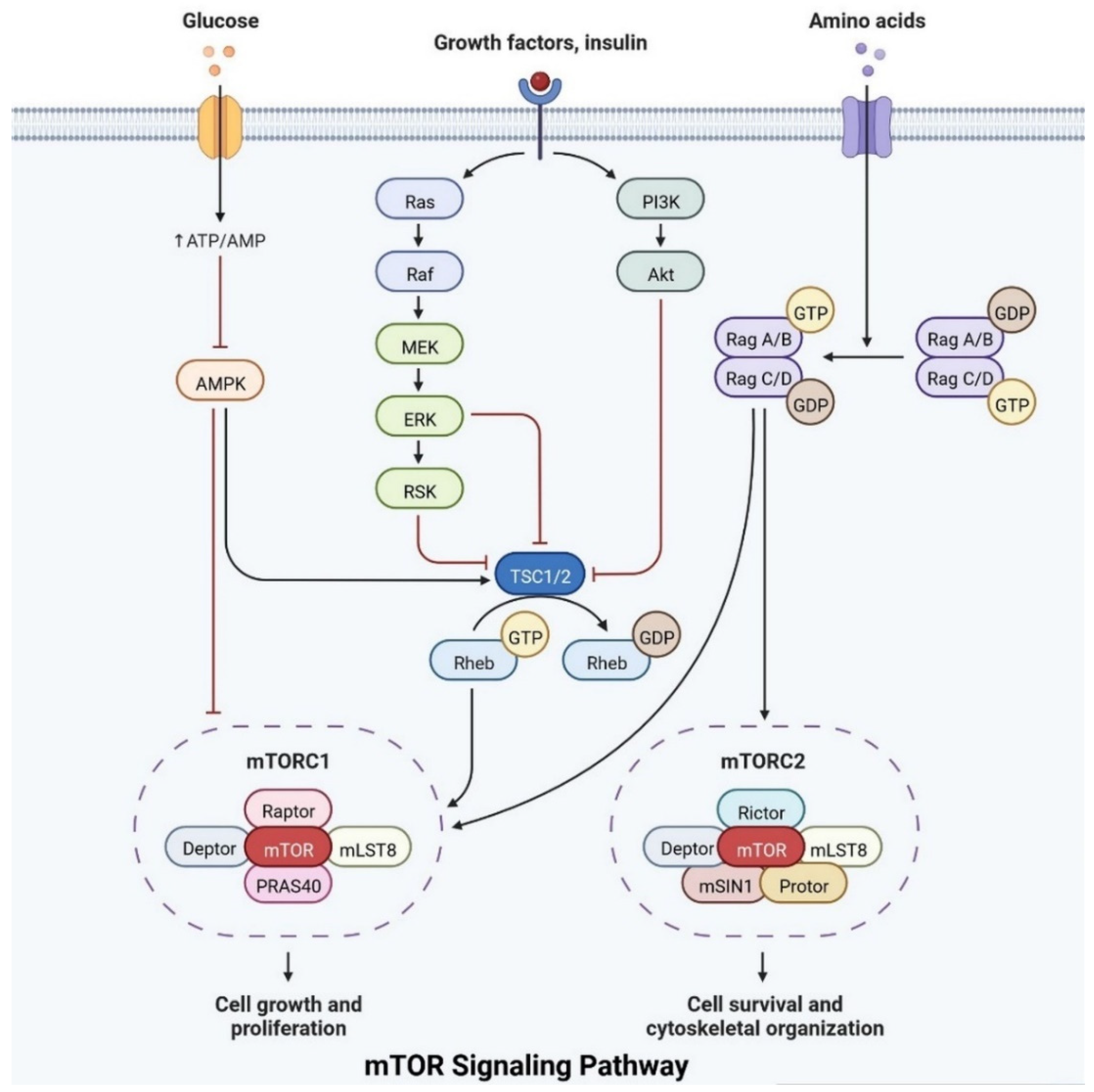
Figure 3. The mTOR pathway integrates growth factor signaling to regulate cellular metabolism, growth, and survival.
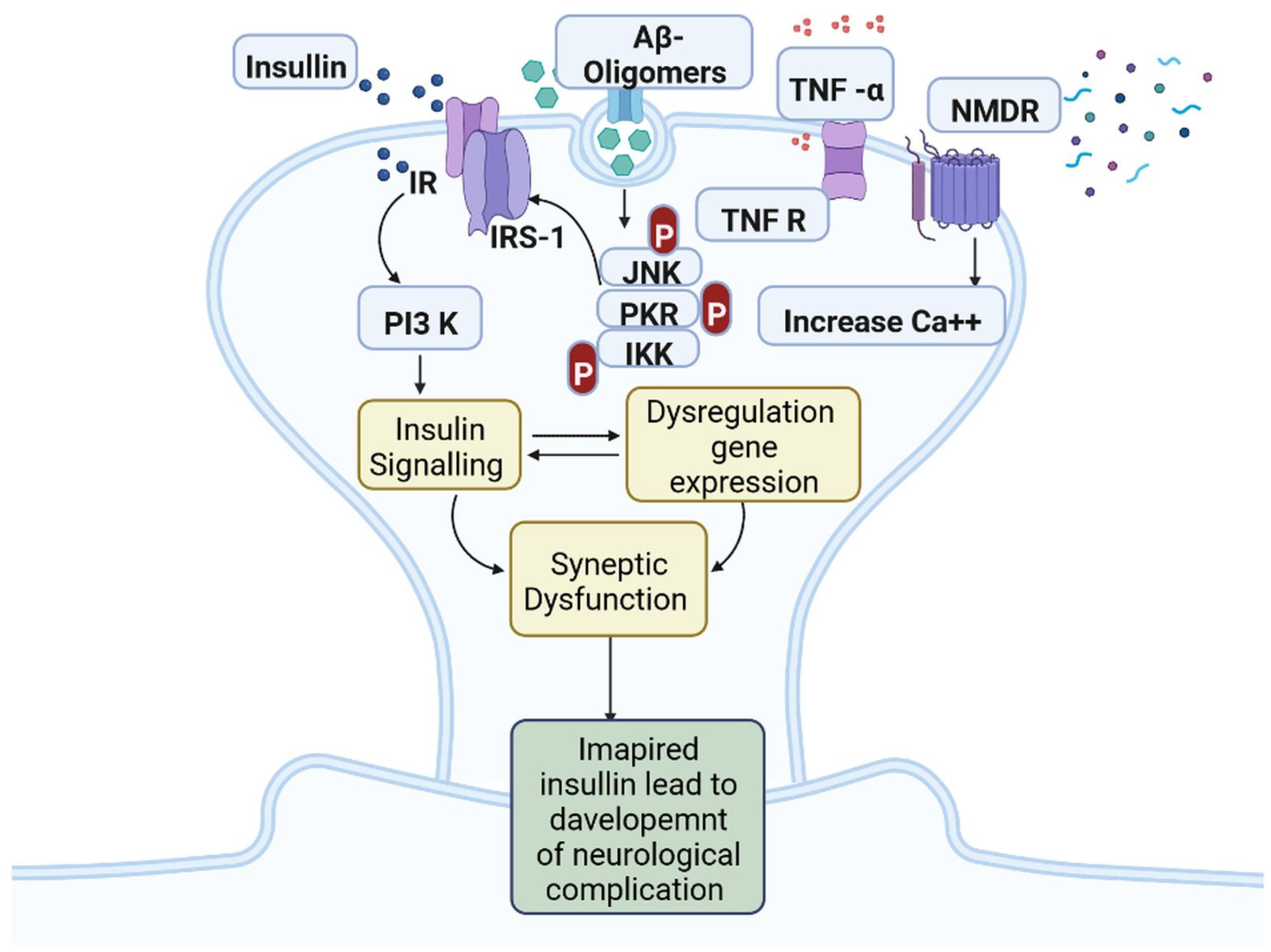
Figure 4. Insulin resistance and development of neuro-complications: the activation of IR autophosphorylation (IRP) leads to the tyrosine phosphorylation of IRS-1 (insulin receptor substrate 1), which activates PI3K (phosphoinositide 3-kinase) and decreases synaptic plasticity and memory.
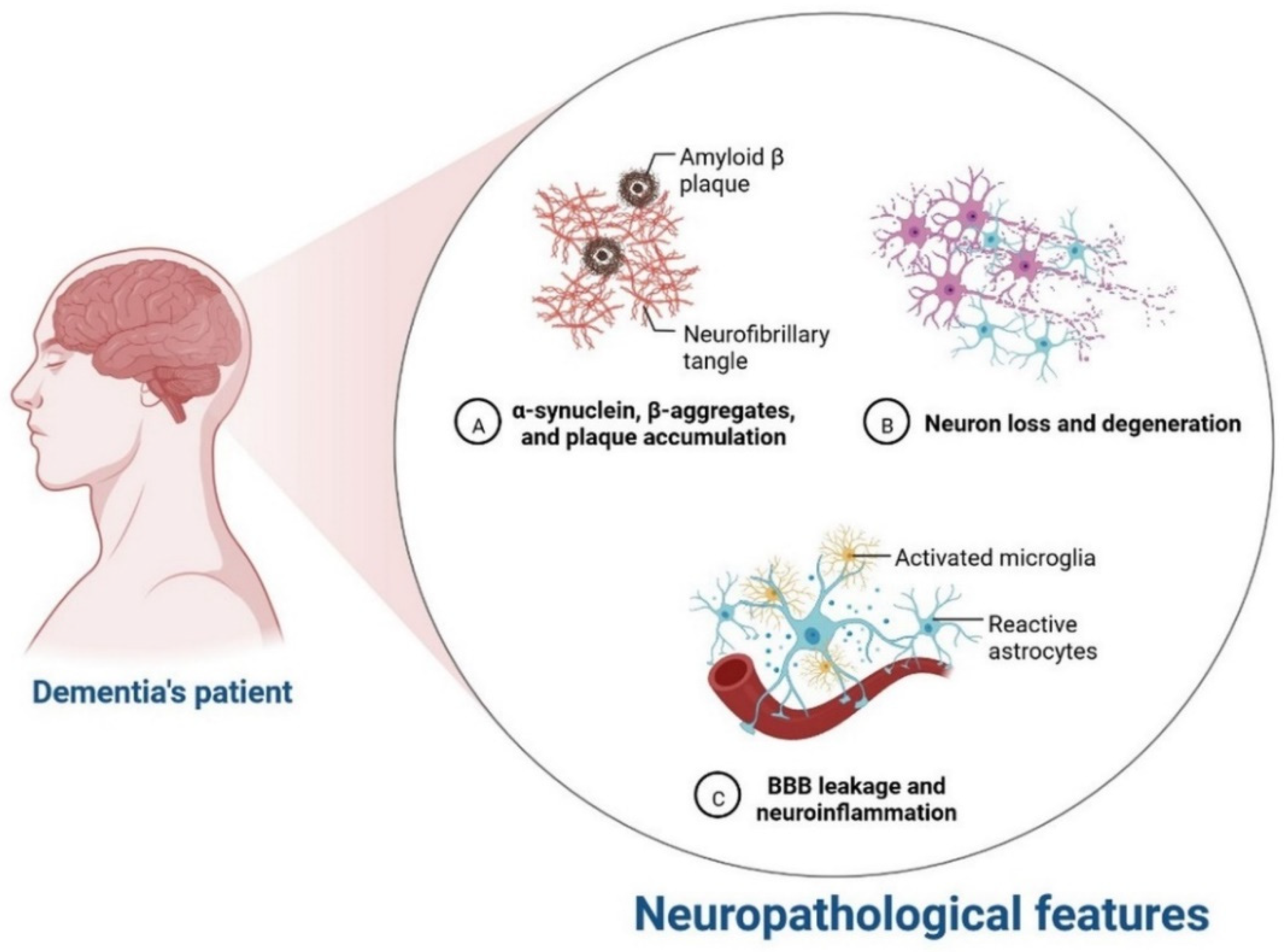
Figure 5. Neurological complication in dementia patients.
3. Relationship between Insulin Receptor m-TOR Pathway
Insulin plays an important role in cell growth, repair, activation, dendritic development, synaptic maintenance, and neuroprotection and is actively involved in learning and memory. Various investigations have shown that changes in insulin levels in the brain lead to the development of neurological complications [11]. Insulin also activates the N-methyl-D-aspartate receptor on the cell membrane, cortical cerebral glucose metabolism, acetylcholine, and norepinephrine, which are actively involved in memory-encoding mechanisms [12]. In the cerebral cortex, IRS (insulin receptor substrate 1) is abundant in the cerebral cortex, hippocampus, hypothalamus, olfactory bulb, septum, and amygdala. Insulin signaling is also involved in regulating synaptic remodeling, which is involved in memory consolidation [13]. T2DM patients have lower brain insulin receptor sensitivity, downregulate the IRS-1, and have lower levels of insulin-like growth factors, as well as lower insulin levels in CSF [14]. Insulin signaling is controlled by mTOR pathways, as shown in Figure 3.
IRs are actively involved in memory and tau phosphorylation, due to the loss of IR receptor activity and downregulated Aβ (Amyloid beta) oligomer binding sites in the synapse [15]. The accumulation of Aβ leads to the development of AD. IGF signaling activation promotes amyloid-protein precursor (APP-A) trafficking, as well as the accumulation of amyloid processing, tau phosphorylation, and a reduction in cerebral blood flow [16]. IRs are transmembrane receptors and are made with alpha and beta subunits. Both the subunits are activated by insulin, which then activates tyrosine kinase enzymes for phosphorylation, which leads to conformational changes in their structure [10]. Changes in their structures favor binding with PI3K (phosphoinositide 3-kinases) and binding with the IRS [13]. After interaction with IRs, the inactive form of PI3K becomes active. The active PI3K enzyme is generated by PIP2 (phosphorylate phosphatidylinositol (4,5)-bisphosphate) in the cell membrane, which then causes the creation of phosphatidylinositol (3,4,5)-trisphosphate (PIP3), which activates AKT/PKB (Figure 3) [17]. Studies on animal models have revealed that IR inhibitors prevent the memory-encoding mechanism. Injecting insulin in an animal model increases the memory-encoding process [17]. IR decreases AKT activity, which inhibits GSK-3 and causes tau protein to be hyperphosphorylated [18]. AKT signaling regulates various responses at the cellular level, such as glycogen synthase kinase-3 beta (GSK3) neuronal survival and TAU phosphorylation [16]. Increasing the synthesis of GSK3β may alter the post-translational modifications in MAPs (microtubule-associated proteins), such as tau protein [16]. Dementia is also caused by mutations in APP (amyloid precursor protein). Synaptic signaling is interrupted by Aβ fibrils infiltrating into synaptic clefts [17]. Polymers of Aβ also play an important role in the development of dementia [10]. The collection of APP alters the ion channel mechanism and disrupts the altered glucose homeostasis, leading to neuronal integrity degradation and cell death [19].
4. Development of Dementia due to Genetic Modifications
mRNA expression and the downregulation of associated receptors are linked to dementia [20]. The oxidative-phosphorylation-related genes are expressed in dementia patients. The mtDNA irregularities are associated with phenotypic variability [21]. Genetic abnormalities are caused by chromosomal defects and damage from neuronal oxidation. The development of dementia due to T2DM is linked with genetic variability [22]. APO E (apolipoprotein E) is expressed by chromosome 19 and exists in three isoforms: apo e2, apo e3, and polymorphic in nature [23]. More than 75 loci have been identified for the development of disease traits [24]. ADAM17 (A disintegrin and metalloprotease 17), ICA1 (Islet Cell Autoantigen 1), DOC2A (Double C2 Domain Alpha), DGKQ (Diacylglycerol Kinase Theta), and ICA1L (Islet Cell Autoantigen 1 Like) are the genes responsible for the regulation of APP metabolism via non-amyloid pathways. Cognitive impairment is caused by HMGB1 (High-mobility group box protein 1), RAGE (Receptor for Advanced Glycation End Products), and TLR4 (Toll-like receptor 4) in hyperglycemic conditions [25]. All these genes impair endothelial cell function and may disrupt various signaling pathways, resulting in an accumulation [26]. Dementia is also associated with the APP, PS1 (presenilin 1), and PS2 genes. Mutations in these genes cause IR in astrocytes and microglial cells [27].
5. Progression of Dementia due to Dopamine Dysregulation in Substantia Nigra
Insulin secretion and glucose homeostasis serve as a basis for neural modulation. Degenerative and functional disorders of the central nervous system are directly related to dementia. Dopamine is a neurotransmitter that plays a major role in neurological complications, as shown in Figure 6.
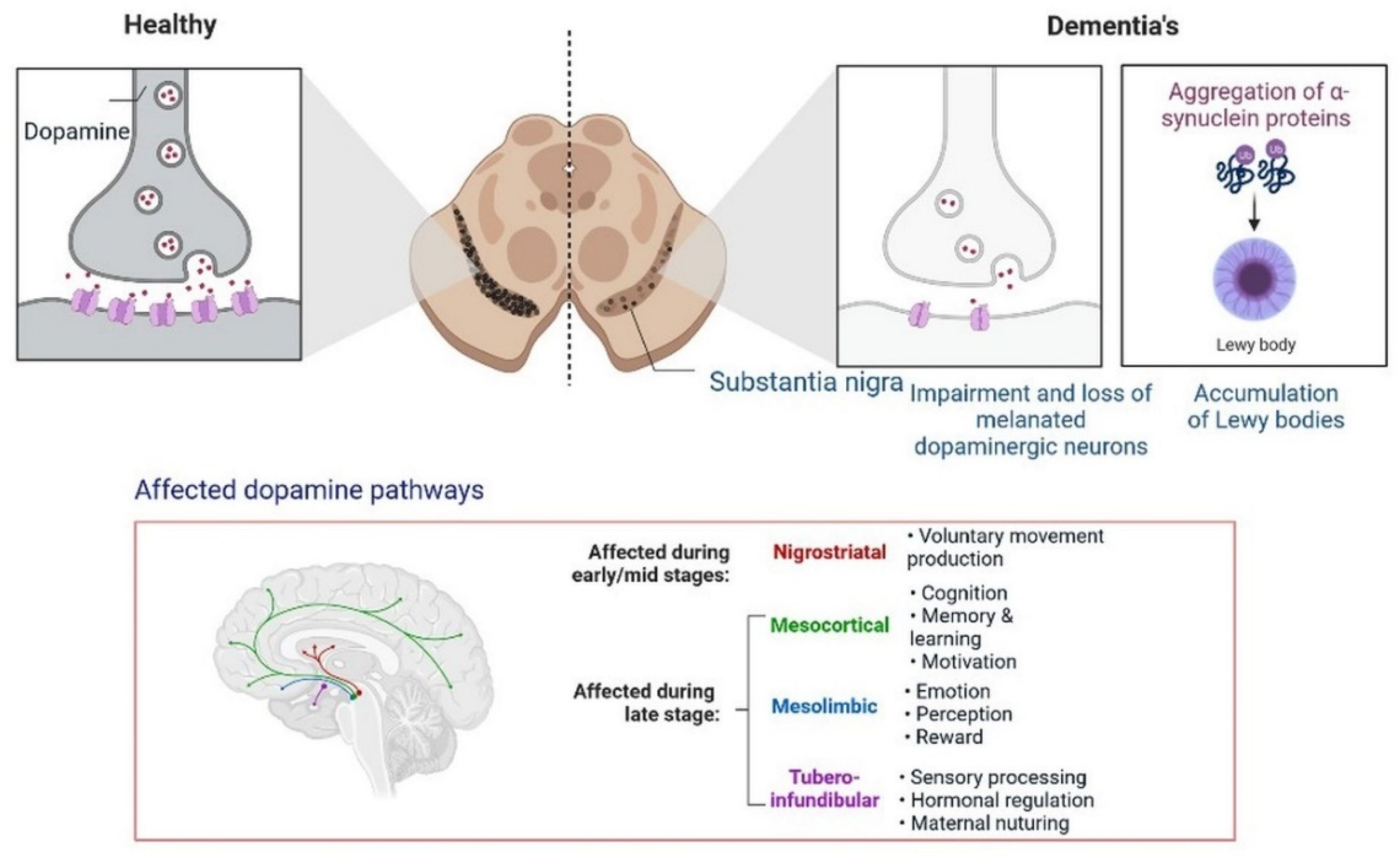
Figure 6. Progression of dementia due to dopamine dysregulation in substantia nigra.
Poor insulin activity in the brain is linked with a high level of cholinergic action, leading to the development of dementia [28].
Furthermore, the development of dementia also occurs due to alterations in the dopamine pathway in the substantia nigra. A PPAR agonist causes memory loss by increasing intracellular glucose oxidation uptake in neurons [29]. Dysregulation in cholinergic neurotransmission alters the performance of the hippocampus area, the recollection of memory, acetylcholine-induced responses, and increased ChE activity in the brain during cognitive deficits. In low amounts, choline acetyltransferase (ChAT) is observed in patients with dementia and neuronal dysfunction [30]. All these findings can be used for identification and the development of etiology and treatment options.
6. Neuronal Apoptosis in Dementia
IR and T2DM result in neuronal death. Different neurological conditions, such as Huntington’s disease, amyotrophic lateral sclerosis and dementia, Parkinson’s disease, and Alzheimer’s disease, may occur because of apoptosis [31], IGF (insulin-like growth factor) [32], increased Bax/Bcl-x ratio, hippocampal neuronal death and L caspase-3 activity, mitochondrial dysfunction, cerebral blood vessel dysfunction, myelin and axon damage, intracytoplasmic calcium deposition, and Purkinje cell damage; IGF-I, IGF-IR, and IR activity; endoplasmic dysfunction, BBB degradation; ependymal [33].
7. Significance of Ketone Bodies in Diabetes-Related Dementia
Brain cells use ketone bodies as a source of energy in situations of nutrient deprivation, after exercise, or low carbohydrates. Regulation of ketone bodies is also linked with gluconeogenesis, the tricarboxylic acid cycle, and fatty acid b-oxidation. Antioxidant responses to ketone bodies are increased [34]. Hyperglycemic conditions can reduce the activity of GABA and glutamate neurotransmitters. Cholinergic transmission was found to be dysregulated in the brain hippocampus [35]. Another neurotransmitter, dopamine, is also associated with behavior, cognition, and emotions. Reduced levels of dopamine receptors have been observed in patients with type 2 diabetes [36]. The 5-HT (5-hydroxytryptamine) neurotransmitters are associated with neuronal cell regeneration and synaptic plasticity. Glucagon-like peptide-1 hormone (GLP-1H) inhibits IR and neuroinflammation in the brain under oxidative stress. GLP1H is also involved in the regulation of synaptic plasticity and neurogenesis [37]. Ketosis increases the levels of GABA (gamma-aminobutyric acid) and excitatory glutamate and regulates the levels of serotonin and dopamine, which are linked with depression and anxiety [38]. Ketone bodies in the hydroxybutyrate form are involved in neuronal anti-apoptosis pathways and cell survival [38]. In the mouse model, fat-rich animals induce APP/PS1xdb/db deposition, whereas ketone bodies improve cognitive impairment function [35]. Ketone bodies regulate neural signaling, increase the sensitivity of insulin, reduce the effects of oxidative stress, increase synaptic activity, and maintain the level of neurotransmitter activity. Low ketone body levels may cause pathological conditions in T2DM [39]. More research is needed to understand the regulation of ketone bodies at low levels in neuroprotection and neurotoxicity at high levels in diabetes-induced dementia treatment options.
8. The Function of Mitochondria in Diabetes-Related Cognitive Impairment
Cell signaling molecules and transcription factors play a very important role in intracellular energy metabolism in the mitochondria. Brain disorders linked with diabetes are caused by abnormalities in mitochondrial functions [40]. IR is also caused by mitochondrial dysfunction, oxidative stress, neuronal damage, decreased mETC (mitochondrial electron transport chain) activity and ATP synthesis, apoptosis, lipid peroxide accumulation, decreased glutathione peroxidase activity, ferroptosis, and An accumulation, all of which can lead to cognitive impairment [41]. Mitophagy is regulated by PINK1 (PTEN-induced kinase 1) and protects the neurons, as has been observed in animal models. PINK1-dependent mitophagy via MT2/Akt/NF-κB is achieved by melatonin, which prevents ROS (reactive oxygen species) accumulation and apoptosis (Figure 7) [42].
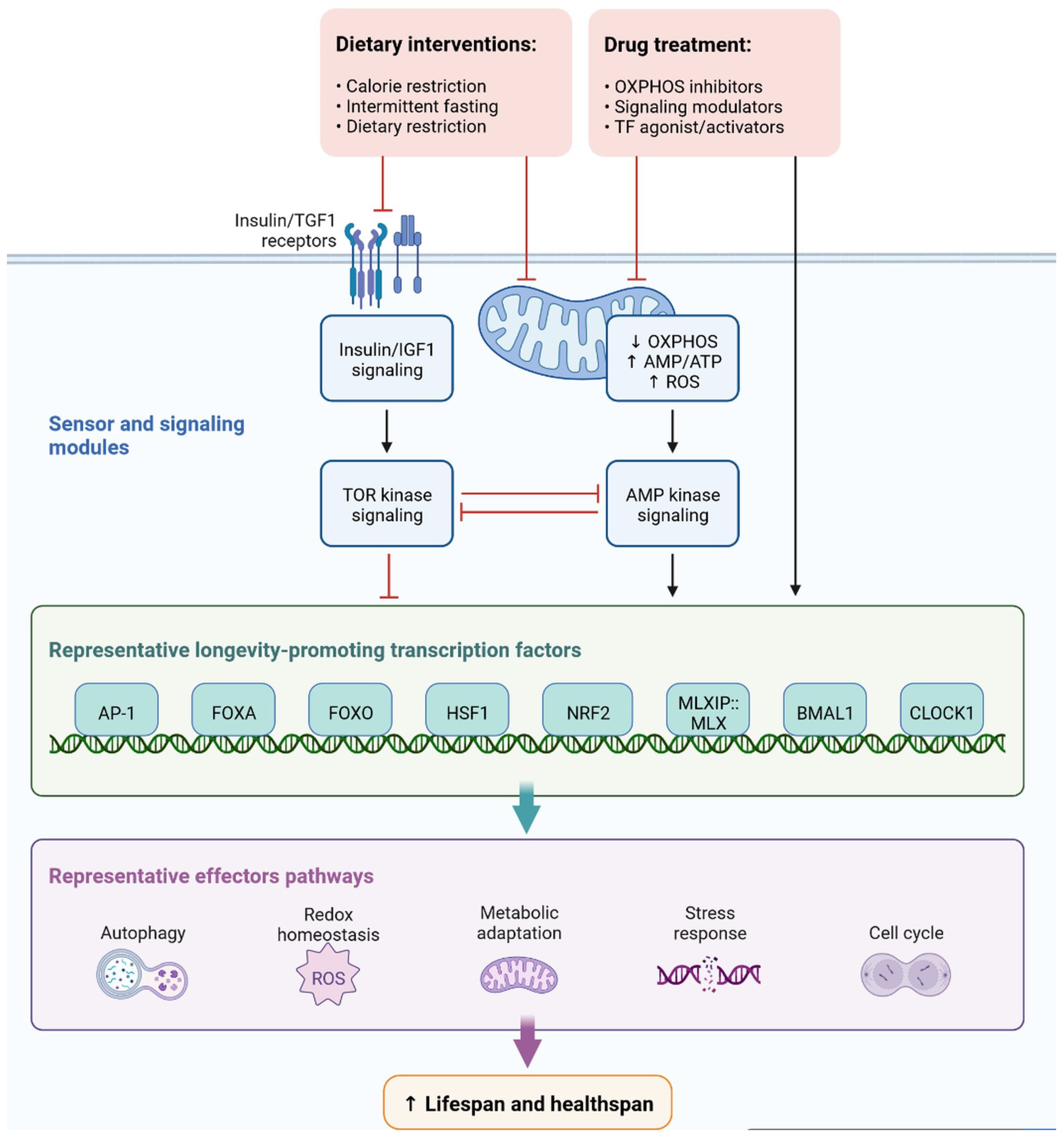
Figure 7. Representation of sensor and signaling molecule dietary intervention and treatment option regulated by effector pathways.
In diabetic mice, increased levels of LC3-II (microtubule-associated protein 1A/1B-light chain 3) and p62 (nucleoporin p62) and decreased levels of PINK1 have been observed, which leads to blocked autophagy [43]. Dephosphorylation of FUNDC1 was also observed in T2DM mice. Cognitive impairment in T2DM mice occurs due to homeostasis, impaired mitophagy, proteostasis disorder, and damage to multiple mechanisms [44].
9. Progressive Dementia Due to Microglial Overactivation
Microglia cells are expressed by the AAβ protein and increase brain insulin levels due to Aβ accumulation. Protein synthesis is being studied as a potential target for drug development and dementia development. Apoptosis occurs due to the generation of ROS and AGE products [45]. Neuroinflammation is also observed due to plaque and tangle formation, oxidative stress markers, oxidized lipids and proteins, and ROS, which is insulin resistant and causes dementia. Microglial cell stimulation and increased levels of proinflammatory cytokines, such as interleukin-1, IL-6, and tumor necrosis factor, inhibit neurogenesis and cause cognitive deficits (Figure 8) [46]. More research is needed to investigate the potential of neuroinflammation treatment, and the cognitive impairment caused by diseases in their early stages.
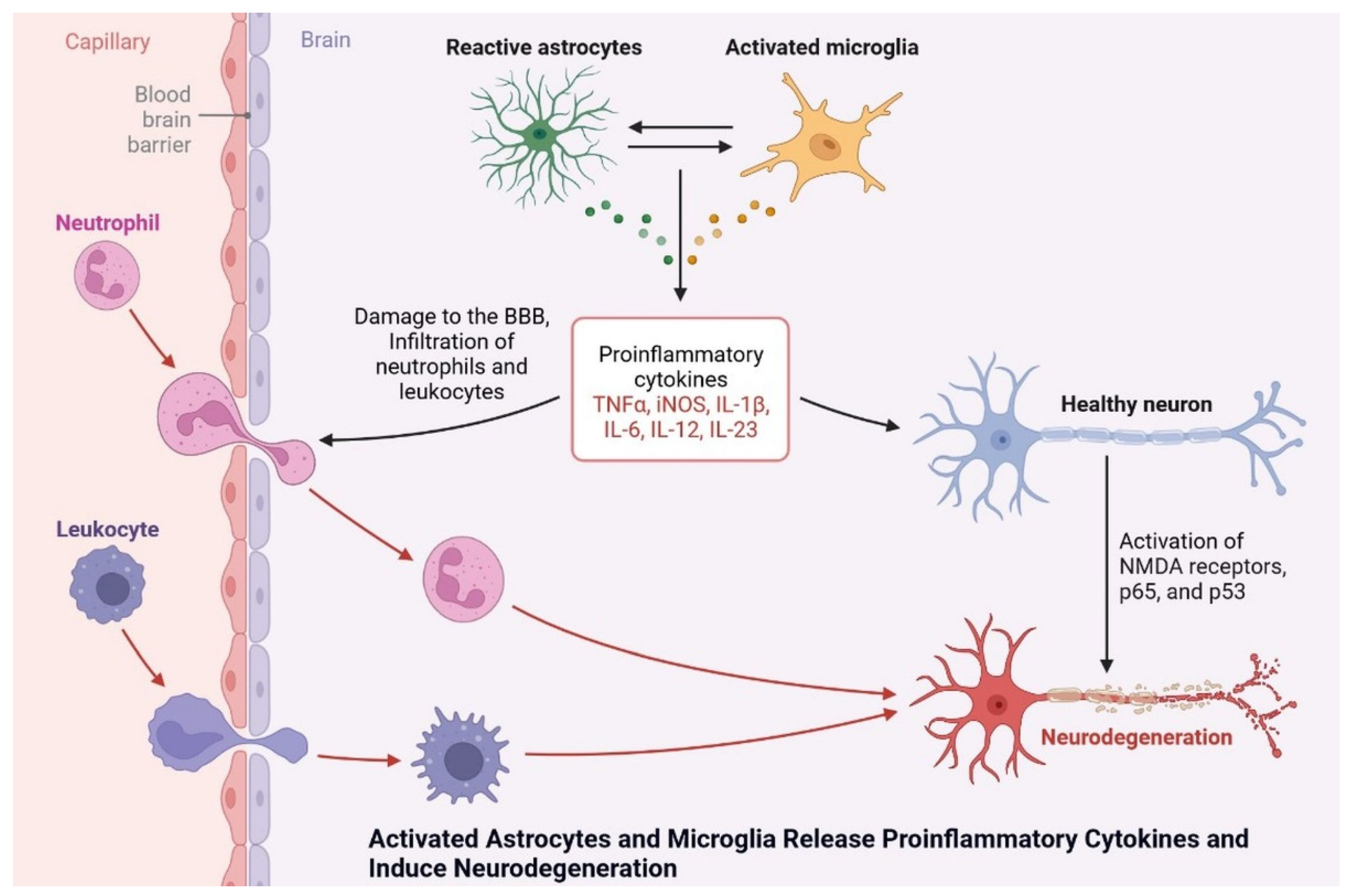
Figure 8. Activated astrocytes and microglia release proinflammatory cytokines and induce neurodegeneration.
References
- Shah, K.; DeSilva, S.; Abbruscato, T. The Role of Glucose Transporters in Brain Disease: Diabetes and Alzheimer’s Disease. Int. J. Mol. Sci. 2012, 13, 12629–12655.
- Vulturar, R.; Chiș, A.; Pintilie, S.; Farcaș, I.M.; Botezatu, A.; Login, C.C.; Sitar-Taut, A.-V.; Orasan, O.H.; Stan, A.; Lazea, C.; et al. One Molecule for Mental Nourishment and More: Glucose Transporter Type 1—Biology and Deficiency Syndrome. Biomedicines 2022, 10, 1249.
- Holman, G.D. Structure, Function and Regulation of Mammalian Glucose Transporters of the SLC2 Family. Pflugers Arch. Eur. J. Physiol. 2020, 472, 1155–1175.
- Koepsell, H. Glucose Transporters in Brain in Health and Disease. Pflugers Arch. Eur. J. Physiol. 2020, 472, 1299–1343.
- Frazier, H.N.; Ghoweri, A.O.; Anderson, K.L.; Lin, R.-L.; Popa, G.J.; Mendenhall, M.D.; Reagan, L.P.; Craven, R.J.; Thibault, O. Elevating Insulin Signaling Using a Constitutively Active Insulin Receptor Increases Glucose Metabolism and Expression of GLUT3 in Hippocampal Neurons. Front. Neurosci. 2020, 14, 668.
- Jurcovicova, J. Glucose Transport in Brain—Effect of Inflammation. Endocr. Regul. 2014, 48, 35–48.
- Thomassen, J.Q.; Tolstrup, J.S.; Benn, M.; Frikke-Schmidt, R. Type-2 Diabetes and Risk of Dementia: Observational and Mendelian Randomisation Studies in 1 Million Individuals. Epidemiol. Psychiatr. Sci. 2020, 29, e118.
- Lyu, F.; Wu, D.; Wei, C.; Wu, A. Vascular Cognitive Impairment and Dementia in Type 2 Diabetes Mellitus: An Overview. Life Sci. 2020, 254, 117771.
- Bell, S.M.; Barnes, K.; de Marco, M.; Shaw, P.J.; Ferraiuolo, L.; Blackburn, D.J.; Venneri, A.; Mortiboys, H. Mitochondrial Dysfunction in Alzheimer’s Disease: A Biomarker of the Future? Biomedicines 2021, 9, 63.
- Oddo, S. The Role of MTOR Signaling in Alzheimer Disease. Front. Biosci. 2012, S4, 941–952.
- Shaughness, M.; Acs, D.; Brabazon, F.; Hockenbury, N.; Byrnes, K.R. Role of Insulin in Neurotrauma and Neurodegeneration: A Review. Front. Neurosci. 2020, 14, 547175.
- Chen, Y.; Deng, Y.; Zhang, B.; Gong, C.-X. Deregulation of Brain Insulin Signaling in Alzheimer’s Disease. Neurosci. Bull. 2014, 30, 282–294.
- Zheng, M.; Wang, P. Role of Insulin Receptor Substance-1 Modulating PI3K/Akt Insulin Signaling Pathway in Alzheimer’s Disease. 3 Biotech 2021, 11, 179.
- Matsuzaki, T.; Sasaki, K.; Tanizaki, Y.; Hata, J.; Fujimi, K.; Matsui, Y.; Sekita, A.; Suzuki, S.O.; Kanba, S.; Kiyohara, Y.; et al. Insulin Resistance Is Associated with the Pathology of Alzheimer Disease: The Hisayama Study. Neurology 2010, 75, 764–770.
- Smolina, K.; Wotton, C.J.; Goldacre, M.J. Risk of Dementia in Patients Hospitalised with Type 1 and Type 2 Diabetes in England, 1998–2011: A Retrospective National Record Linkage Cohort Study. Diabetologia 2015, 58, 942–950.
- Hobday, A.L.; Parmar, M.S. The Link Between Diabetes Mellitus and Tau Hyperphosphorylation: Implications for Risk of Alzheimer’s Disease. Cureus 2021, 13, e18362.
- Heras-Sandoval, D.; Avila-Muñoz, E.; Arias, C. The Phosphatidylinositol 3-Kinase/MTor Pathway as a Therapeutic Target for Brain Aging and Neurodegeneration. Pharmaceuticals 2011, 4, 1070–1087.
- Yang, L.; Wang, H.; Liu, L.; Xie, A. The Role of Insulin/IGF-1/PI3K/Akt/GSK3β Signaling in Parkinson’s Disease Dementia. Front. Neurosci. 2018, 12, 73.
- Hu, Z.; Jiao, R.; Wang, P.; Zhu, Y.; Zhao, J.; de Jager, P.; Bennett, D.A.; Jin, L.; Xiong, M. Shared Causal Paths Underlying Alzheimer’s Dementia and Type 2 Diabetes. Sci. Rep. 2020, 10, 4107.
- Canchi, S.; Raao, B.; Masliah, D.; Rosenthal, S.B.; Sasik, R.; Fisch, K.M.; de Jager, P.L.; Bennett, D.A.; Rissman, R.A. Integrating Gene and Protein Expression Reveals Perturbed Functional Networks in Alzheimer’s Disease. Cell Rep. 2019, 28, 1103–1116.e4.
- Ramos-Campoy, O.; Lladó, A.; Bosch, B.; Ferrer, M.; Pérez-Millan, A.; Vergara, M.; Molina-Porcel, L.; Fort-Aznar, L.; Gonzalo, R.; Moreno-Izco, F.; et al. Differential Gene Expression in Sporadic and Genetic Forms of Alzheimer’s Disease and Frontotemporal Dementia in Brain Tissue and Lymphoblastoid Cell Lines. Mol. Neurobiol. 2022, 59, 6411–6428.
- Wang, X.; Lopez, O.L.; Sweet, R.A.; Becker, J.T.; DeKosky, S.T.; Barmada, M.M.; Demirci, F.Y.; Kamboh, M.I. Genetic Determinants of Disease Progression in Alzheimer’s Disease. J. Alzheimer’s Dis. 2014, 43, 649–655.
- Liu, C.-C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer Disease: Risk, Mechanisms and Therapy. Nat. Rev. Neurol. 2013, 9, 106–118.
- Dove, A.; Shang, Y.; Xu, W.; Grande, G.; Laukka, E.J.; Fratiglioni, L.; Marseglia, A. The Impact of Diabetes on Cognitive Impairment and Its Progression to Dementia. Alzheimer’s Dement. 2021, 17, 1769–1778.
- Bottero, V.; Potashkin, J.A. Meta-Analysis of Gene Expression Changes in the Blood of Patients with Mild Cognitive Impairment and Alzheimer’s Disease Dementia. Int. J. Mol. Sci. 2019, 20, 5403.
- Newcombe, E.A.; Camats-Perna, J.; Silva, M.L.; Valmas, N.; Huat, T.J.; Medeiros, R. Inflammation: The Link between Comorbidities, Genetics, and Alzheimer’s Disease. J. Neuroinflammation 2018, 15, 276.
- Hiltunen, M.; Khandelwal, V.K.M.; Yaluri, N.; Tiilikainen, T.; Tusa, M.; Koivisto, H.; Krzisch, M.; Vepsäläinen, S.; Mäkinen, P.; Kemppainen, S.; et al. Contribution of Genetic and Dietary Insulin Resistance to Alzheimer Phenotype in APP/PS1 Transgenic Mice. J. Cell. Mol. Med. 2012, 16, 1206–1222.
- Duarte, A.I.; Moreira, P.I.; Oliveira, C.R. Insulin in Central Nervous System: More than Just a Peripheral Hormone. J. Aging Res. 2012, 2012, 384017.
- Iannotti, F.A.; Vitale, R.M. The Endocannabinoid System and PPARs: Focus on Their Signaling Crosstalk, Action and Transcriptional Regulation. Cells 2021, 10, 586.
- Chen, Z.-R.; Huang, J.-B.; Yang, S.-L.; Hong, F.-F. Role of Cholinergic Signaling in Alzheimer’s Disease. Molecules 2022, 27, 1816.
- De Mello, N.P.; Orellana, A.M.; Mazucanti, C.H.; de Morais Lima, G.; Scavone, C.; Kawamoto, E.M. Insulin and Autophagy in Neurodegeneration. Front. Neurosci. 2019, 13, 491.
- Al-Samerria, S.; Radovick, S. The Role of Insulin-like Growth Factor-1 (IGF-1) in the Control of Neuroendocrine Regulation of Growth. Cells 2021, 10, 2664.
- O’Kusky, J.; Ye, P. Neurodevelopmental Effects of Insulin-like Growth Factor Signaling. Front. Neuroendocrinol. 2012, 33, 230–251.
- Jensen, N.J.; Wodschow, H.Z.; Nilsson, M.; Rungby, J. Effects of Ketone Bodies on Brain Metabolism and Function in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8767.
- Chung, J.Y.; Kim, O.Y.; Song, J. Role of Ketone Bodies in Diabetes-Induced Dementia: Sirtuins, Insulin Resistance, Synaptic Plasticity, Mitochondrial Dysfunction, and Neurotransmitter. Nutr. Rev. 2022, 80, 774–785.
- Juárez Olguín, H.; Calderón Guzmán, D.; Hernández García, E.; Barragán Mejía, G. The Role of Dopamine and Its Dysfunction as a Consequence of Oxidative Stress. Oxidative Med. Cell. Longev. 2016, 2016, 9730467.
- Kim, Y.-K.; Kim, O.Y.; Song, J. Alleviation of Depression by Glucagon-Like Peptide 1 Through the Regulation of Neuroinflammation, Neurotransmitters, Neurogenesis, and Synaptic Function. Front. Pharmacol. 2020, 11, 1270.
- McGrath, T.; Baskerville, R.; Rogero, M.; Castell, L. Emerging Evidence for the Widespread Role of Glutamatergic Dysfunction in Neuropsychiatric Diseases. Nutrients 2022, 14, 917.
- Dilliraj, L.N.; Schiuma, G.; Lara, D.; Strazzabosco, G.; Clement, J.; Giovannini, P.; Trapella, C.; Narducci, M.; Rizzo, R. The Evolution of Ketosis: Potential Impact on Clinical Conditions. Nutrients 2022, 14, 3613.
- Galizzi, G.; Di Carlo, M. Insulin and Its Key Role for Mitochondrial Function/Dysfunction and Quality Control: A Shared Link between Dysmetabolism and Neurodegeneration. Biology 2022, 11, 943.
- De Felice, F.G.; Ferreira, S.T. Inflammation, Defective Insulin Signaling, and Mitochondrial Dysfunction as Common Molecular Denominators Connecting Type 2 Diabetes to Alzheimer Disease. Diabetes 2014, 63, 2262–2272.
- Shan, Z.; Fa, W.H.; Tian, C.R.; Yuan, C.S.; Jie, N. Mitophagy and Mitochondrial Dynamics in Type 2 Diabetes Mellitus Treatment. Aging 2022, 14, 2902–2919.
- Potenza, M.A.; Sgarra, L.; Desantis, V.; Nacci, C.; Montagnani, M. Diabetes and Alzheimer’s Disease: Might Mitochondrial Dysfunction Help Deciphering the Common Path? Antioxidants 2021, 10, 1257.
- Luo, J.-S.; Ning, J.-Q.; Chen, Z.-Y.; Li, W.-J.; Zhou, R.-L.; Yan, R.-Y.; Chen, M.-J.; Ding, L.-L. The Role of Mitochondrial Quality Control in Cognitive Dysfunction in Diabetes. Neurochem. Res. 2022, 47, 2158–2172.
- Ahmad, R.; Chowdhury, K.; Kumar, S.; Irfan, M.; Reddy, G.S.; Akter, F.; Jahan, D.; Haque, M. Diabetes Mellitus: A Path to Amnesia, Personality, and Behavior Change. Biology 2022, 11, 382.
- Selman, A.; Burns, S.; Reddy, A.P.; Culberson, J.; Reddy, P.H. The Role of Obesity and Diabetes in Dementia. Int. J. Mol. Sci. 2022, 23, 9267.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.4K
Entry Collection:
Nitric Oxide: Physiology, Pharmacology, and Therapeutic Applications
Revisions:
2 times
(View History)
Update Date:
30 Nov 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No