Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Markus Karl Muellner | -- | 2588 | 2022-11-29 18:28:50 | | | |
| 2 | Peter Tang | Meta information modification | 2588 | 2022-11-30 03:03:00 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Radhakrishnan, S.; Hoff, O.; Muellner, M.K. Bivalent Proximity-Inducing Compounds for Targeted Protein Degradation. Encyclopedia. Available online: https://encyclopedia.pub/entry/37170 (accessed on 09 August 2026).
Radhakrishnan S, Hoff O, Muellner MK. Bivalent Proximity-Inducing Compounds for Targeted Protein Degradation. Encyclopedia. Available at: https://encyclopedia.pub/entry/37170. Accessed August 09, 2026.
Radhakrishnan, Sridhar, Oskar Hoff, Markus K. Muellner. "Bivalent Proximity-Inducing Compounds for Targeted Protein Degradation" Encyclopedia, https://encyclopedia.pub/entry/37170 (accessed August 09, 2026).
Radhakrishnan, S., Hoff, O., & Muellner, M.K. (2022, November 29). Bivalent Proximity-Inducing Compounds for Targeted Protein Degradation. In Encyclopedia. https://encyclopedia.pub/entry/37170
Radhakrishnan, Sridhar, et al. "Bivalent Proximity-Inducing Compounds for Targeted Protein Degradation." Encyclopedia. Web. 29 November, 2022.
Copy Citation
Bivalent proximity-inducing compounds represent a novel class of small molecule therapeutics with exciting potential and new challenges. The most prominent examples of such compounds are utilized in targeted protein degradation where E3 ligases are hijacked to recruit a substrate protein to the proteasome via ubiquitination.
PROTAC
PIC
targeted protein degradation
proximity-inducing compound
E3 recruiter
ubiquitination
E3 ligase
1. Introduction
While the domain of small-molecule drugs has largely been the space of competitive inhibitors, a novel class of compounds with enormous potential for therapeutics has emerged within recent years. These new “proximity-inducing compounds” (PICs) combine approaches from protein–protein inhibitor (PPi) development with the hijacking of protein degradation machinery or other functional modules within the cell by creating proximity to a substrate protein. In many instances this proximity can then be sufficient to trigger the desired cellular response such as the addition of post-translational modifications or translocation. Targeted protein degradation (TPD) has become a promising therapeutic modality for tackling harmful proteins using a fundamentally novel mechanism of action for small molecules. In the case of bivalent proximity-inducing degraders (Figure 1)—often also termed PROTACs (proteolysis-targeting chimera), CIP (chemical inducer of proximity) or degronomid—a molecule recruits an E3 ligase (or other protein tied to degradation machinery) to a target protein of interest (POI). This results in ternary complex formation between the POI, the degrader and the E3 ligase, which can lead to ubiquitination and degradation by the proteasome. The degrader is released in the process of degradation and is then free to start another degradation cycle.
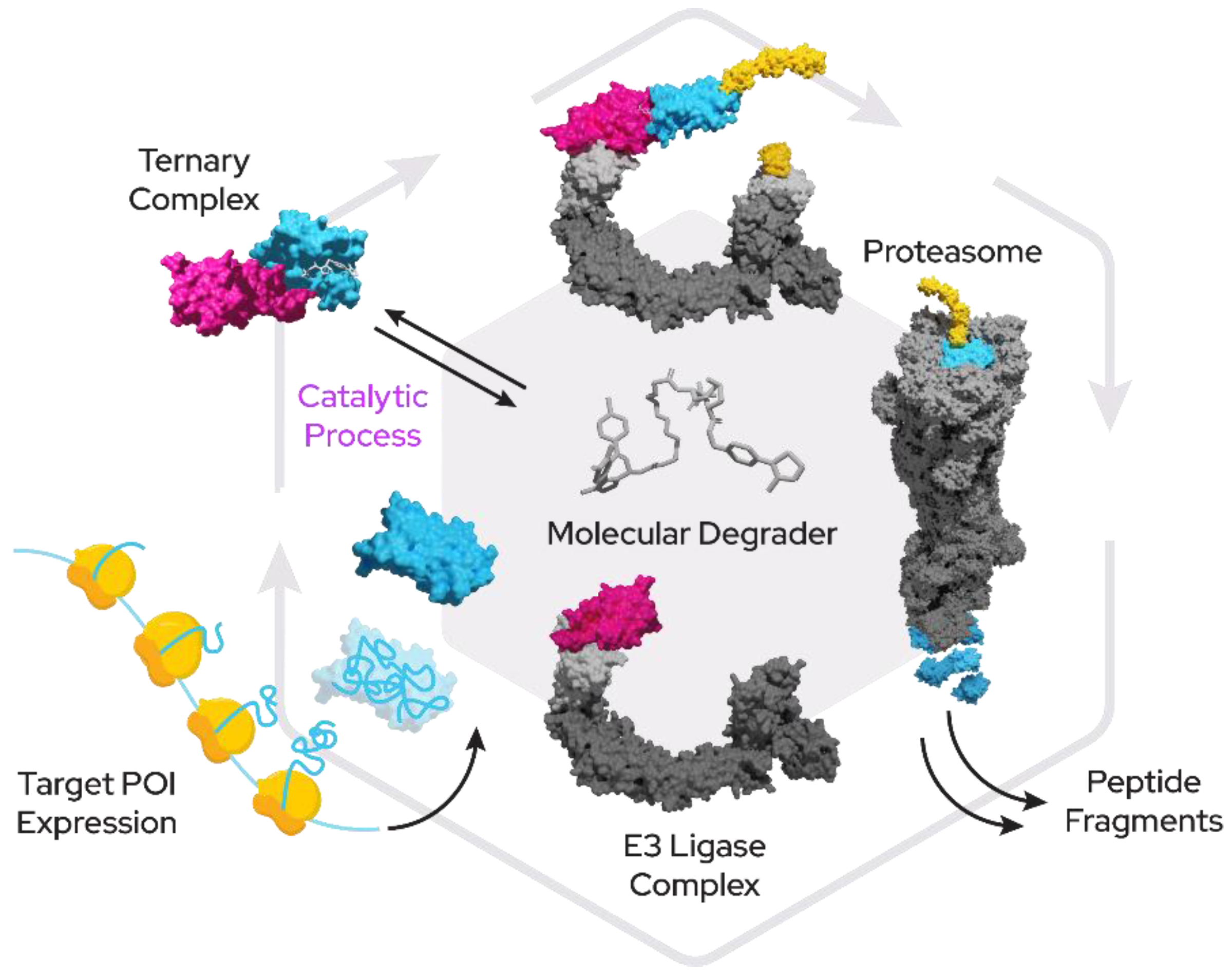
Figure 1. Proximity-inducing compounds inducing targeted protein degradation. A bivalent molecular degrader forms a ternary complex with an E3 ligase (pink) and a target protein (blue) which leads to attachment of ubiquitin chains (yellow) and subsequent proteasomal degradation.
Monovalent degraders on the other hand bind only onto one protein and modify its surface properties to enable binding of degradation machinery.
Finally, the third and current generation of companies uses machine learning and AI as tools to quickly and more efficiently arrive at optimized drug-like orally bioavailable degrader designs (Figure 2).
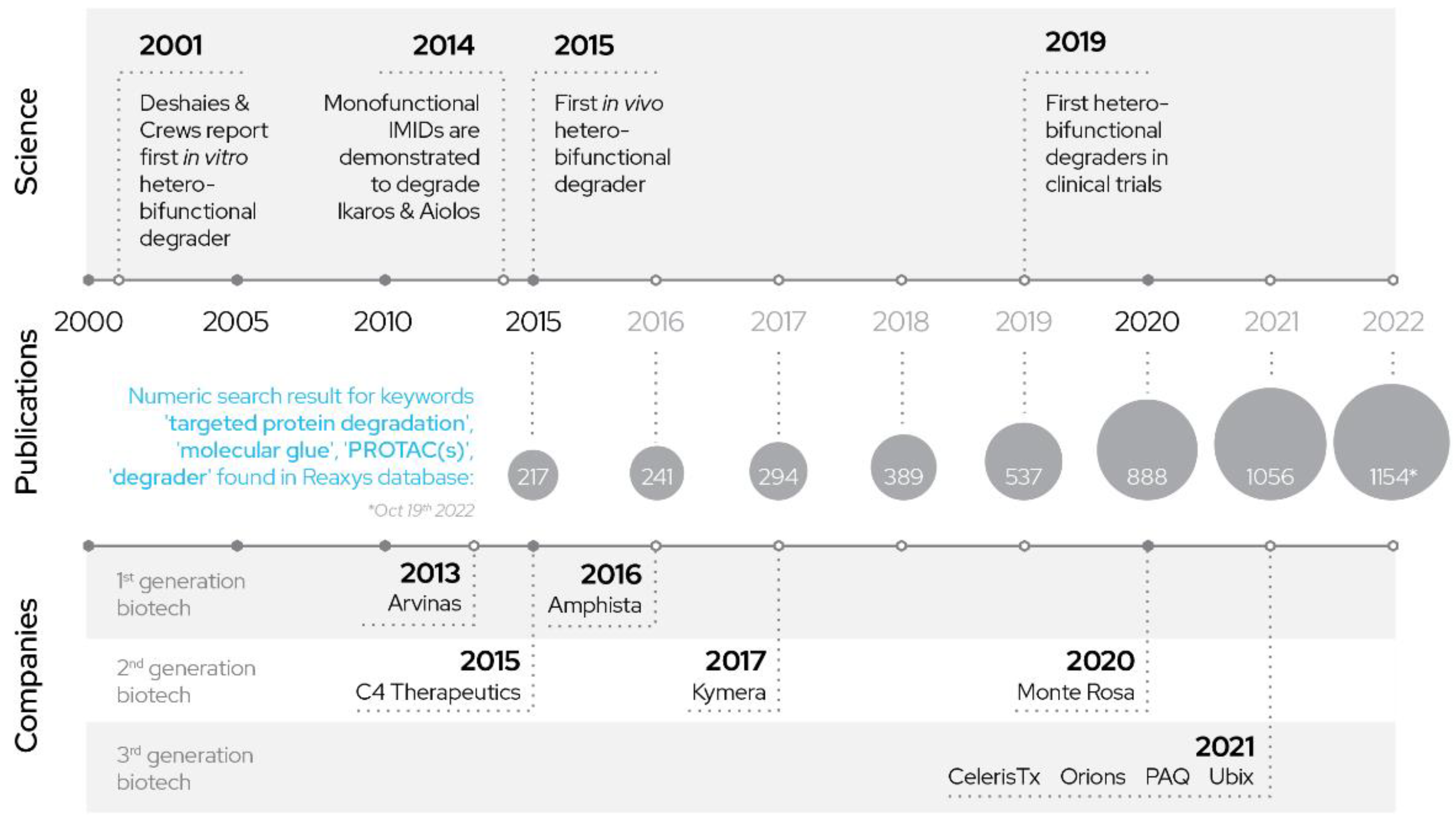
Figure 2. History of PROTACs: development of PROTACs over years.
2. Prominent E3 Ligases in Targeted Protein Degradation
Under physiological conditions, E3 ligases represent an integral part of the ubiquitin proteasomal system by providing specific substrate recognition, often dependent on modifications of specific residues on the target protein, for instance by enzymatic activity.
A prime example here is the well-studied HIF1α oxygen-dependent degradation domain (ODD) that, after oxygen-dependent hydroxylation by prolyl hydroxylases, is recognized by the von Hippel–Lindau tumor suppressor gene (VHL) and subsequently leads to degradation of HIF1α through formation of a complex of Elongin B, Elongin C and the Cullin 2 proteins [1][2].
The family of E3 ligases represents a large untapped pool of potential for targeted protein degradation and modulation of protein function. Only a handful of these ligases have so far been successfully used in the context of developing proximity-inducing compounds and fewer still have a ligand with favorable properties. Based on the presence of protein domains, there are an estimated 500–1000 E3s in human cells, yet for the vast majority not much is known about their target proteins, degrons or even cellular function. Prominent exceptions are ligases frequently mutated in cancer such as MDM2, FBXW7, VHL, KEAP1 and BRCA1, as well as E3 ligases found in genome-wide association studies and phenotypic screens (for instance Parkin, ITCH) as well as serendipitous discoveries such as CRBN as a target of thalidomide [3].
The first PROTAC (recognized as such) was a fusion between the compound ovalicin (binding MetAP-2) and an IκB-α phosphopeptide that binds to the E3 ligase β-TRCP, which was able to recruit the SCF complex to MetAP-2 and lead to degradation [4].
Several other targets were linked and subsequently degraded using this approach; however, the peptidic nature of the recruiter created several issues for more advanced drug discovery.
Likewise, the HIF1α recognition sequence for VHL (briefly mentioned above) was shown to be useful as a recruiter for VHL in targeted protein degradation. While the peptide itself proved difficult to use in drug development, it was later successfully developed into a peptidomimetic compound with more favorable properties and now represents one of the two most-used recruiters in the TPD space [5].
The recruiter for CRBN, among the most often used recruiters to date, was found in a much more serendipitous (and tragic) manner. The severe teratogenic side-effects of a popular drug developed by Grünenthal in the 1950s, Contergan (thalidomide), led to an investigation into the mechanism of action and discovered that thalidomide, among other targets, binds to the E3 ligase CRBN as well as SALL4-most likely responsible for the developmental defects [6], (p. 4), [7].
Since thalidomide was a non-peptidic recruiter of an E3 ligase with favorable properties (aside from the teratogenicity the compound was remarkably non-toxic), thalidomide and its derivatives lenalidomide and pomalidomide soon became attractive components in degrader design. Georg Winter and Jay Bradner’s 2015 science paper [8] then ultimately clearly demonstrated the potential of CRBN-based degraders and moved targeted protein degradation from a mostly academic endeavor to a new therapeutic modality.
Another E3 recruiter was born of the significant effort concentrated on the discovery of therapeutic approaches to prevent TP53 inactivation, one of the most frequent events in cancer. These studies led to the discovery of the E3 ligase and proto-oncogene MDM2. MDM2 acts as an E3 ligase that can target TP53 for degradation and is amplified in several types of cancer. Drug discovery efforts here led to the successful development of PPi inhibitors for TP53:MDM2 interaction-namely the compound nutlin and its derivatives.
Similarly, the E3 ligase KEAP1 and its protein target NRF2 represent a frequently altered pathway in oncology and, again, significant efforts were made to find PPi inhibitors as therapeutic agents, which culminated in several candidates to be later repurposed in targeted protein degradation approaches.
3. Classification of E3 Ligases
Based on structural and functional features (Figure 3), currently exploited E3 ligases can roughly be divided into two categories:
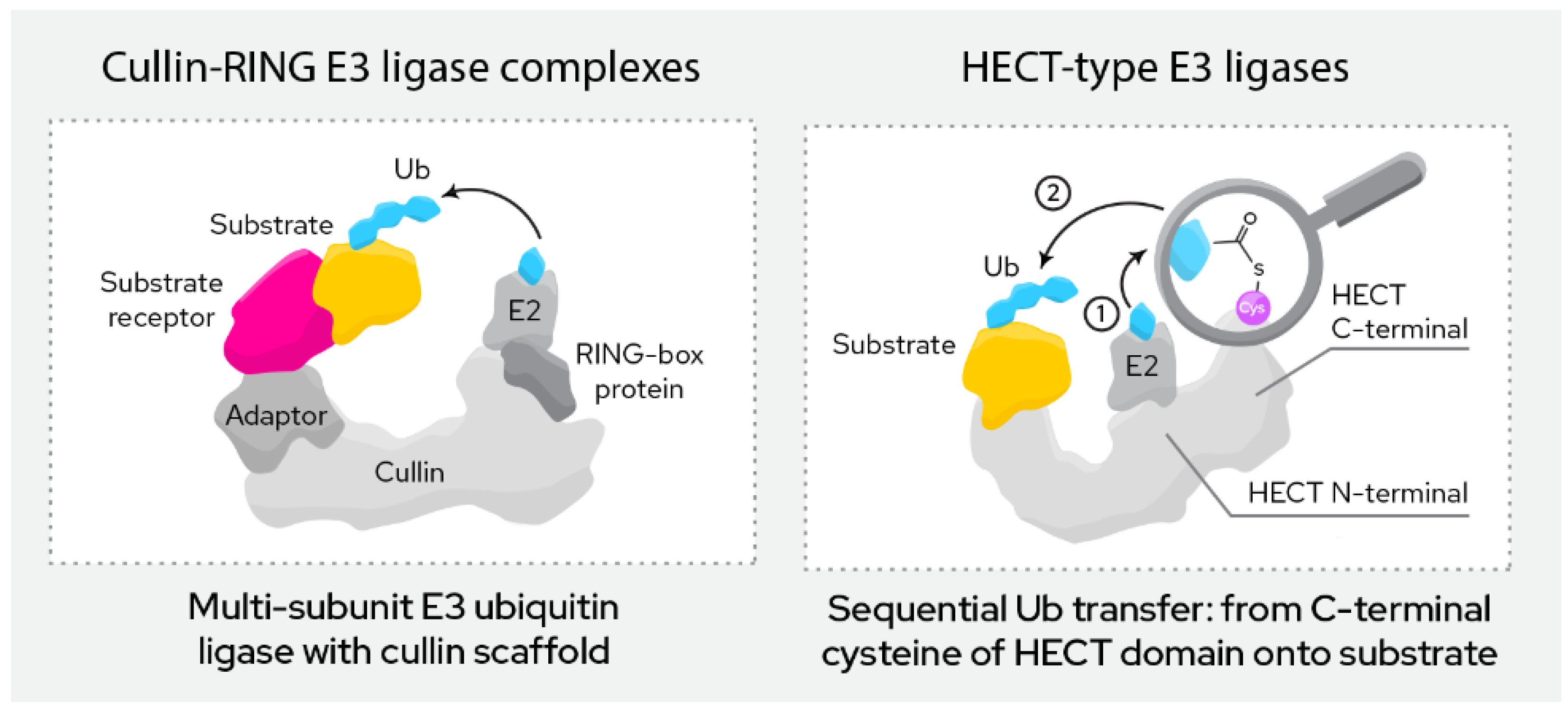
Figure 3. Most relevant classes of E3 ligases in TPD.
3.1. RING Finger E3 Ligases
This group of ligases is defined by their RING (“really interesting new gene”) domain. This domain binds to the E2 conjugating enzyme in the course of ubiquitination, which then directly transfers the ubiquitin onto the target protein.
RING finger E3 ligases can be further divided into monomeric and multi-subunit ligases. Monomeric RING ligases often also auto-ubiqutinate while multi-subunit ligases are assembled from a larger number of proteins that form the active complex. Among the most common are the Cullin-RING ligases (CRL), the APC/C ligases and SCF ligase complexes. Multi-subunit ligases are typically highly regulated by post-translational modifications. The ligases discussed below (CRBN, VHL, MDM2, cIAP, XIAP, KEAP1, DCAF16, DCAF15, DCAF11 and RNF4) all belong to this class.
3.2. HECT E3 Ligases
This group of ligases all carry a HECT domain (“homologous to the E6AP carboxyl terminus”), to which the E2 ligases transfer the ubiquitin as an intermediary step before the ubiquitin is then transferred onto the target protein. HECT ligases can be further divided into the NEDD4 family, the HERC family and a more heterogeneous group of remaining members that do not fit either of the previous two.
3.3. Other Ligases
While the above present the prominent E3 ligase families, others exist. For instance, in a group of ligases called U-Box ligases, named after the distinctive U-box domain, the E2 transfers the ubiquitin directly onto the substrate similar to the RING ligases.
The RBR (RING-IBR-RING) ligase family represents a mix between RING and HECT E3 ligases. They contain two RING domains (RING1 and RING2) but also, like HECT ligases, transfer the ubiquitin first onto the E3 and then subsequently onto the target, rather than directly from the E2. Of note, the linkage of ubiquitin in the poly-ubiquitin chain here is performed in a linear fashion by the LUBAC complex though methionine linkage rather than the more common linkage through one of the seven lysines of ubiquitin.
Increasing the space of employable E3 ligases for TPD is of enormous interest due to limitations in available recruiters and their properties (discussed in detail below), limited freedom to operate due to a confined intellectual property space when it comes to degrader design as well as untapped potential for tissue-specific drugs by using E3 expression in the tissue of interest to avoid unwanted off-target effects (Figure 4.) [9].
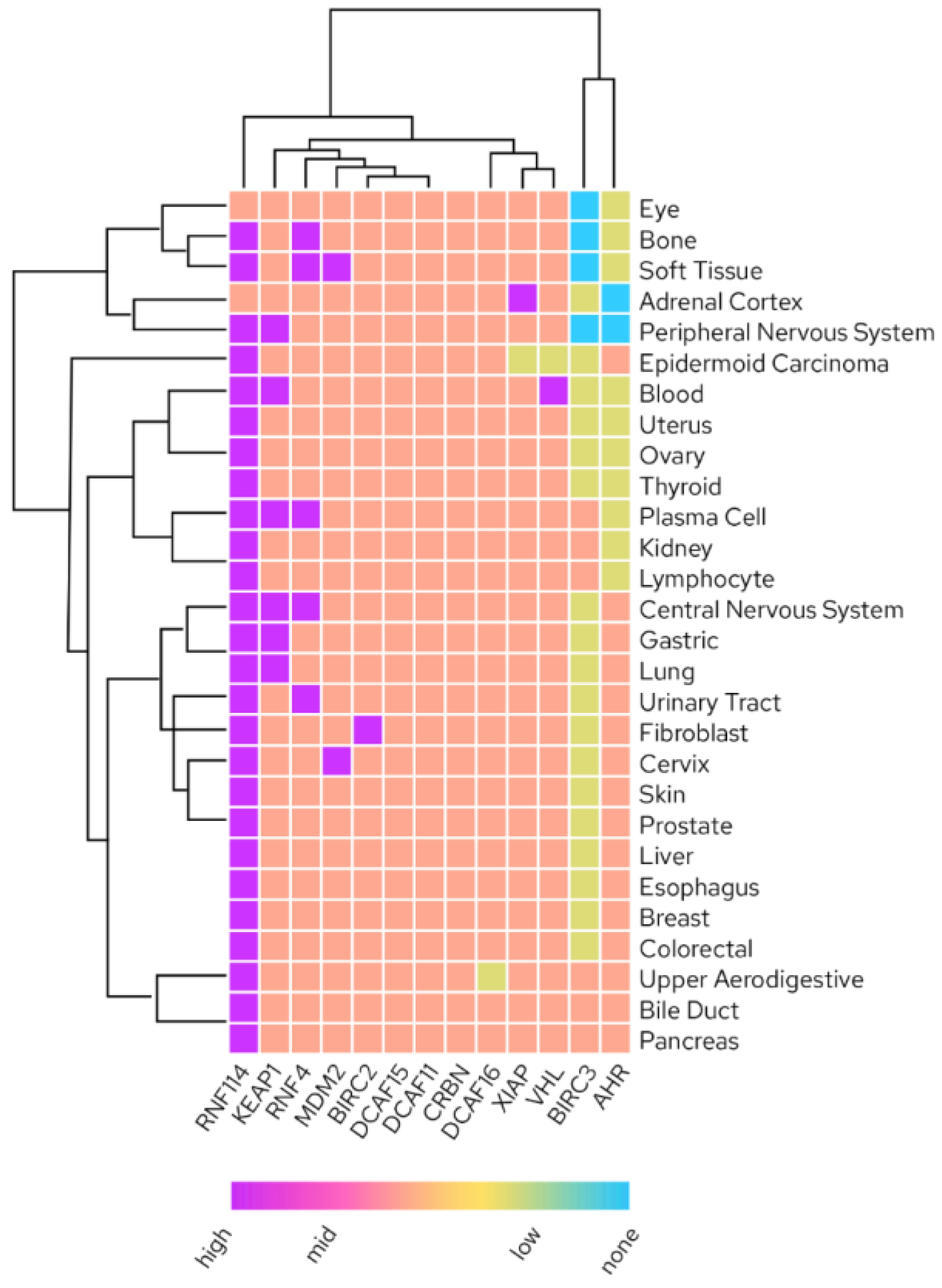
Figure 4. Tissue RNA expression of liganded E3 ligases (Human Protein Atlas; proteinatlas.org accessed on 1 October 2022).
4. Most Prominent Recruiters in TPD
While VHL and CRBN represent the majority of E3 ligases regularly utilized for targeted protein degradation, MDM2 and cIAP are also frequently employed. Other E3 ligases have been liganded (detailed below) but are still in earlier development.
4.1. CRBN
Thalidomide, lenalidomide and pomalidomide are the common IMiD drugs that can be used as recruiters of the cereblon E3 ligase. It was shown that the glutarimide of the IMiDs goes into the binding site of CRBN and interacts [9][10] strongly with Trp380 and His378 (Figure 5a). Mutations of Trp380 resulted in no binding and phthalimides without glutarimide also showed no binding to CRBN [11]. Furthermore, methylation of the glutarimide moiety in heterobifunctional degraders reduced binding with CRBN and degradation of BRD4. Thus, the glutarimide was shown to be essential for CRBN recruitment. The phenyl ring of the IMiDs is pointing out of the cereblon binding site and is perfectly suited for exit vectors to conjugate target binding warheads through a linker or even directly.
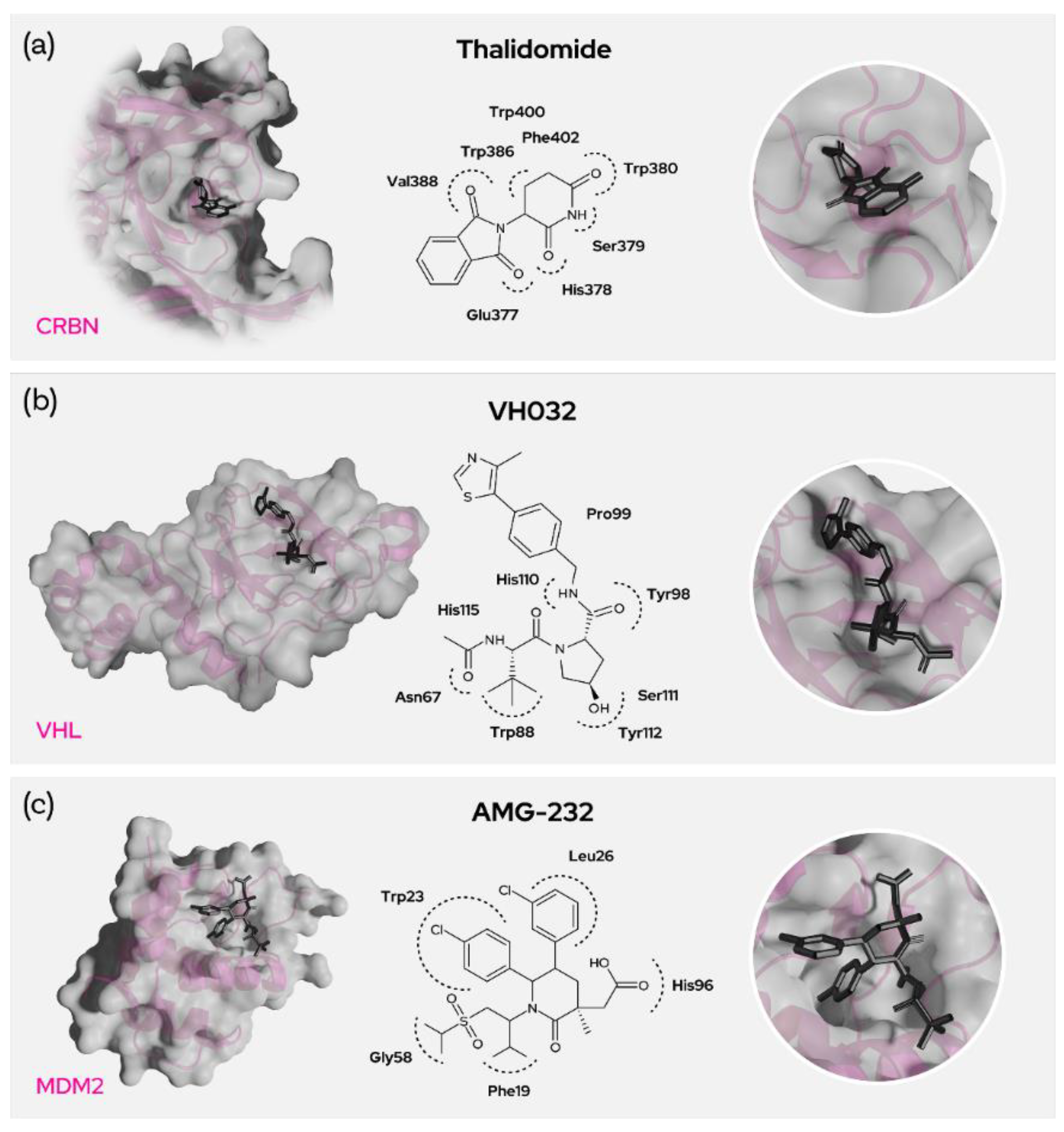
Figure 5. Prominent recruiters and binding residues. (a) Interacting residues between CRBN and Thalidomide, (b) VH032 and VHL as well as (c) AMG-232 and MDM2.
Recently, several CRBN-recruiting degraders were developed using phenyl glutarimides where glutarimides are directly attached to the aryl rings and then conjugated with target warheads [12]. CRBN-based degraders are small in size considering their molecular weight and tPSA, and they have lower cLogP and improved solubility. While adding linker and warhead comes with a gain in size, these degraders often still have better physicochemical properties compared with the VHL-, IAP- or MDM2-recruiting degraders. The relative hydrophilicity of CRBN recruiters not only facilitates solubility but also reduces non-specific interactions/plasma protein binding-thus improving bioavailability.
4.2. VHL
Von Hippel-Lindau (VHL) is also a heavily researched and promising E3 ligase in the development of proximity-induced degraders [13]. The most commonly used recruiter of VHL, VH032, mostly comprises five main fragments (Figure 5b). The thiazole part of the recruiter seems to be necessary, though other substituents seem possible and the methyl substitution at the 4-position of the thiazole ring seems to be pointing outside the E3 binding pocket. This allows for the use of these residues as exit vectors for degrader design.
The phenyl group adjacent to the thiazole is not being extensively explored, but the phenolic replacement is widely known and the ethers with the phenolic group are often used conjugate the linkers carrying the warhead [14]. The hydroxyproline part of the recruiter plays a crucial role in binding with the ligase and the hydroxy group appears buried in the protein site. Epimers here were shown to be inactive, demonstrating that the stereochemistry is unalterable. But there seems to be a fluoro-substitution possibility in place of the hydroxyl group [15] and the possibility of capping the OH to generate pro-drug esters [9]. Stereochemistry is crucial for the amine/amide functional end of the molecule and many substitutions were employed here as this part is the most commonly used exit vector. Though the branched benzylamino fragment does not seem crucial and remains unmodified even with unsubstituted benzylamine, it can be used as an exit vector with the desired stereochemistry for warhead conjugation as it is pointing out of the binding domain. VHL is a promising E3 ligase; however, the binding site is unfortunately rather hydrophobic.
The resulting heterobifunctional degraders, thus mostly hydrophobic as well, might therefore interact with plasma proteins through non-specific interactions and eventually result in poor bioavailability. VHL degraders are usually higher in molecular weight, topological polar surface area and hydrophobicity and hence need to be highly potent to degrade the target protein within the limits of their reduced bioavailability.
4.3. MDM2
E3 ubiquitin-protein ligase MDM2 mediates ubiquitination and subsequent degradation of p53 and inhibits p53-mediated cell cycle arrest and apoptosis. MDM2 acts as an E3 ligase for itself and promotes proteasomal degradation. MDM2 also facilitates proteasomal degradation of DYRK2, IGF1R and SNAI1. MDM2 inhibitors trigger loss of mitochondrial membrane potential, caspase activation and DNA fragmentation. MDM2 is involved in normal hematopoiesis and its inhibition can lead to hematopoietic defects. The main toxicity observed by using MDM2 inhibitors are gastrointestinal and bone marrow [16] disorders. MDM2 is highly expressed in most tissues as well as in several cancers, likely due to its role as a proto-oncogene. Nutlin-based binders imidazole, pyrrolidine and piperidinone (AMG-232) bind to MDM2 amino acid residues Trp23 (p-chloro phenyl goes into the groove and binds), Leu26, His96 (with the acetic acid), Phe19 and Gly58. Nutlins have a central imidazoline ring with two p-chloro phenyl rings and a third phenyl ring with alkoxy groups (Figure 5c). The N1 of the imidazole connects to the piperzine ring via a urea linkage. The compound idasanutlin has a pyrrolidine ring replacing the central imidazole ring, and AMG-232 on the other hand has piperidine ring. All these inhibitor derivatives have the room for linker attachment at the piperazine or phenyl carboxylic acid, which points out of the MDM2 binding site. As an exemplary target using an MDM2 degrader strategy, BCR-ABL was degraded by recruitment of MDM2 through nutlin by means of an alkoxy linker. [17][18]. In general, the MDM2 recruiters have greater molecular weight and lipophilicity, and this further increases upon the degrader designs. However, the recruiters have low tPSA and this also contributes to the relatively lower tPSA of MDM2 ligase-recruiting degraders.
4.4. cIAP1
Cellular inhibitor of apoptosis 1 (c-IAP1), also known as Baculoviral IAP repeat-containing protein 2 (BIRC2), regulates caspases and apoptosis and modulates inflammatory signaling and immunity. IAP family proteins contain a 70 amino acid motif referred to as the baculovirus IAP repeat (BIR) domain and there are three such BIR domains along with ubiquitin binding and RING E3 ligase activity domains (Figure 6a). It functions as an E3 ligase regulating the canonical NF-kappa-B signaling pathway and suppressing the non-canonical NF-kappa-B. The target proteins for its E3 ubiquitin-protein ligase activity include a number of RIP/MAP kinases [19] (p. 2). Specific and nongenetic IAP-dependent protein erasers (SNIPERs) induce degradation of the target protein by forming the ternary complex between the cIAP1 BIR domain and the target protein, which then facilitates ubiquitination of the target with cIAP1-bound E2. Several Ala-Val-Pro-containing SMAC mimetics have been explored as the binders of IAPs. Most of the binders except bestatin have proline analogues as a fixed backbone while bestatin features a serine amino acid instead [20]. Some recruiters have a six-membered nitrogen heterocycle or a fused nitrogen heterocycle replacing the proline.
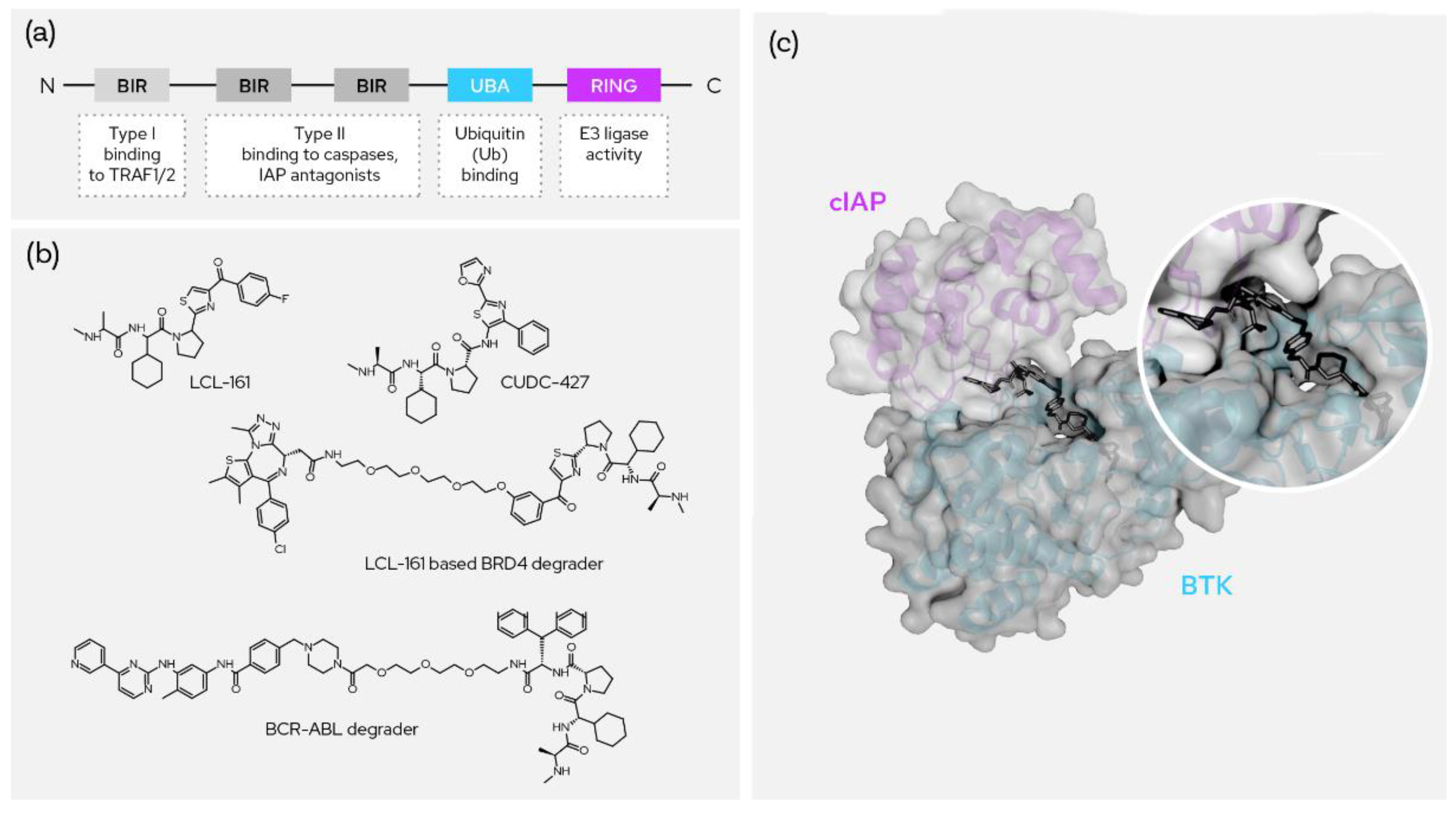
Figure 6. SNIPERs and XIAP-based degraders. (a) The domain structure of cIAP; (b) the two most prominent recruiters and two published degrader molecules using them; (c) ternary crystal structure between molecular degrader, cIAP and BTK (PDB: 6W70).
4.5. XIAP
X-linked inhibitor of apoptosis protein (XIAP) also regulates caspases and apoptosis, inflammatory signaling and immunity, copper homeostasis, mitogenic kinase signaling and cell proliferation. It acts as an E3 ligase in regulating NF-kappa-B signaling and the target proteasome-degraded proteins include RIP kinases and caspases. Piperazine remains at the central core for binding and the indole group is included as a mean for conjugating the target warhead and for capping. One prominent example of a successful XIAP-based degrader was used for the degradation of BCR–ABL using dasatinib as the warhead [21].
References
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha Targeted for VHL-Mediated Destruction by Proline Hydroxylation: Implications for O2 Sensing. Science 2001, 292, 464–468.
- Jaakkola, P.; Mole, D.R.; Tian, Y.-M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau Ubiquitylation Complex by O2-Regulated Prolyl Hydroxylation. Science 2001, 292, 468–472.
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a Primary Target of Thalidomide Teratogenicity. Science 2010, 327, 1345–1350.
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559.
- Klein, V.G.; Townsend, C.E.; Testa, A.; Zengerle, M.; Maniaci, C.; Hughes, S.J.; Chan, K.-H.; Ciulli, A.; Lokey, R.S. Understanding and Improving the Membrane Permeability of VH032-Based PROTACs. ACS Med. Chem. Lett. 2020, 11, 1732–1738.
- Krönke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide Causes Selective Degradation of IKZF1 and IKZF3 in Multiple Myeloma Cells. Science 2014, 343, 301–305.
- Banik, S.M.; Pedram, K.; Wisnovsky, S.; Ahn, G.; Riley, N.M.; Bertozzi, C.R. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 2020, 584, 291–297.
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381.
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419.
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25.
- Akuffo, A.A.; Alontaga, A.Y.; Metcalf, R.; Beatty, M.S.; Becker, A.; McDaniel, J.M.; Hesterberg, R.S.; Goodheart, W.E.; Gunawan, S.; Ayaz, M.; et al. Ligand-mediated protein degradation reveals functional conservation among sequence variants of the CUL4-type E3 ligase substrate receptor cereblon. J. Biol. Chem. 2018, 293, 6187–6200.
- Min, J.; Mayasundari, A.; Keramatnia, F.; Jonchere, B.; Yang, S.W.; Jarusiewicz, J.; Actis, M.; Das, S.; Young, B.; Slavish, J.; et al. Phenyl-Glutarimides: Alternative Cereblon Binders for the Design of PROTACs. Angew. Chem. Int. Ed. 2021, 60, 26663–26670.
- Galdeano, C.; Gadd, M.S.; Soares, P.; Scaffidi, S.; Van Molle, I.; Birced, I.; Hewitt, S.; Dias, D.M.; Ciulli, A. Structure-Guided Design and Optimization of Small Molecules Targeting the Protein–Protein Interaction between the von Hippel–Lindau (VHL) E3 Ubiquitin Ligase and the Hypoxia Inducible Factor (HIF) Alpha Subunit with in Vitro Nanomolar Affinities. J. Med. Chem. 2014, 57, 8657–8663.
- Testa, A.; Hughes, S.J.; Lucas, X.; Wright, J.E.; Ciulli, A. Structure-Based Design of a Macrocyclic PROTAC. Angew. Chem. Int. Ed. 2020, 59, 1727–1734.
- Testa, A.; Lucas, X.; Castro, G.V.; Chan, K.-H.; Wright, J.E.; Runcie, A.C.; Gadd, M.S.; Harrison, W.T.A.; Ko, E.-J.; Fletcher, D.; et al. 3-Fluoro-4-hydroxyprolines: Synthesis, Conformational Analysis, and Stereoselective Recognition by the VHL E3 Ubiquitin Ligase for Targeted Protein Degradation. J. Am. Chem. Soc. 2018, 140, 9299–9313.
- Fang, Y.; Liao, G.; Yu, B. Small-molecule MDM2/X inhibitors and PROTAC degraders for cancer therapy: Advances and perspectives. Acta Pharm. Sin. B 2020, 10, 1253–1278.
- Wurz, R.P.; Cee, V.J. Targeted Degradation of MDM2 as a New Approach to Improve the Efficacy of MDM2-p53 Inhibitors. J. Med. Chem. 2019, 62, 445–447.
- Nietzold, F.; Rubner, S.; Berg, T. The hydrophobically-tagged MDM2–p53 interaction inhibitor Nutlin-3a-HT is more potent against tumor cells than Nutlin-3a. Chem. Commun. 2019, 55, 14351–14354.
- McComb, S.; Cheung, H.H.; Korneluk, R.G.; Wang, S.; Krishnan, L.; Sad, S. cIAP1 and cIAP2 limit macrophage necroptosis by inhibiting Rip1 and Rip3 activation. Cell Death Differ. 2012, 19, 1791–1801.
- Bricelj, A.; Steinebach, C.; Kuchta, R.; Gütschow, M.; Sosič, I. E3 Ligase Ligands in Successful PROTACs: An Overview of Syntheses and Linker Attachment Points. Front. Chem. 2021, 9, 707317.
- Demizu, Y.; Shibata, N.; Hattori, T.; Ohoka, N.; Motoi, H.; Misawa, T.; Shoda, T.; Naito, M.; Kurihara, M. Development of BCR-ABL degradation inducers via the conjugation of an imatinib derivative and a cIAP1 ligand. Bioorganic Med. Chem. Lett. 2016, 26, 4865–4869.
More
Information
Subjects:
Chemistry, Medicinal
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.9K
Revisions:
2 times
(View History)
Update Date:
30 Nov 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No