 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | manlio ferrarini | -- | 6459 | 2022-11-29 11:47:20 | | | |
| 2 | Dean Liu | -3 word(s) | 6456 | 2022-11-30 03:36:45 | | | | |
| 3 | Dean Liu | -5 word(s) | 6451 | 2022-12-01 07:28:40 | | |
Video Upload Options
The engagement of the B cell receptor (BcR) on the surface of leukemic cells represents a key event in chronic lymphocytic leukemia (CLL) since it can lead to the maintenance and expansion of the neoplastic clone. This notion was initially suggested by observations of the CLL BcR repertoire and of correlations existing between certain BcR features and the clinical outcomes of single patients. Based on these observations, tyrosine kinase inhibitors (TKIs), which block BcR signaling, have been introduced in therapy with the aim of inhibiting CLL cell clonal expansion and of controlling the disease. Indeed, the impressive results obtained with these compounds provided further proof of the role of BcR in CLL.
1. Introduction
Lymphoproliferative disorders of mature T and B cells are generally believed to originate from cells that have completed their maturation process. This is unlike what has been observed for most cancers, in which the transforming events occur primarily in the stem cells, which, because of these events, become cancer stem cells, capable of proliferating and of differentiating in part into mature neoplastic cells, with low proliferative capacities [1]. This genesis is possible since mature T and B cells can be recruited into the cell cycle via external signals and propagate the transforming events to their progeny, as stem cells of other tissues do. The stimulation of mature T and B cells by antigens may represent one of the signals which facilitates the process of malignant transformation. Gastric lymphomas are accompanied by Helicobacter pylori (HP) infection, which promotes the formation of lymphoid tissue in the gastric mucosa and the proliferation of B cells that already carry transforming mutations or which cause the accumulation of transforming events while proliferating [2]. Antibiotic treatment results in a regression of the lymphoid gastric lesions, concomitant with the eradication of the HP infection in the early disease stages, whereas it is ineffective or partially effective at later stages, indicating the role of additional transforming events in disease progression [3]. In lymphomas arising in patients with hepatitis C virus (HCV) infection, the BcR of the malignant clone frequently has specificity for HCV epitopes and treatment of the viral infection can result in lymphoma regression [4][5]. In CLL, the analysis of the repertoire indicates the involvement of BcR in promoting clonal expansion.
2. BcR Signaling in CLL
2.1. Tonic Signals
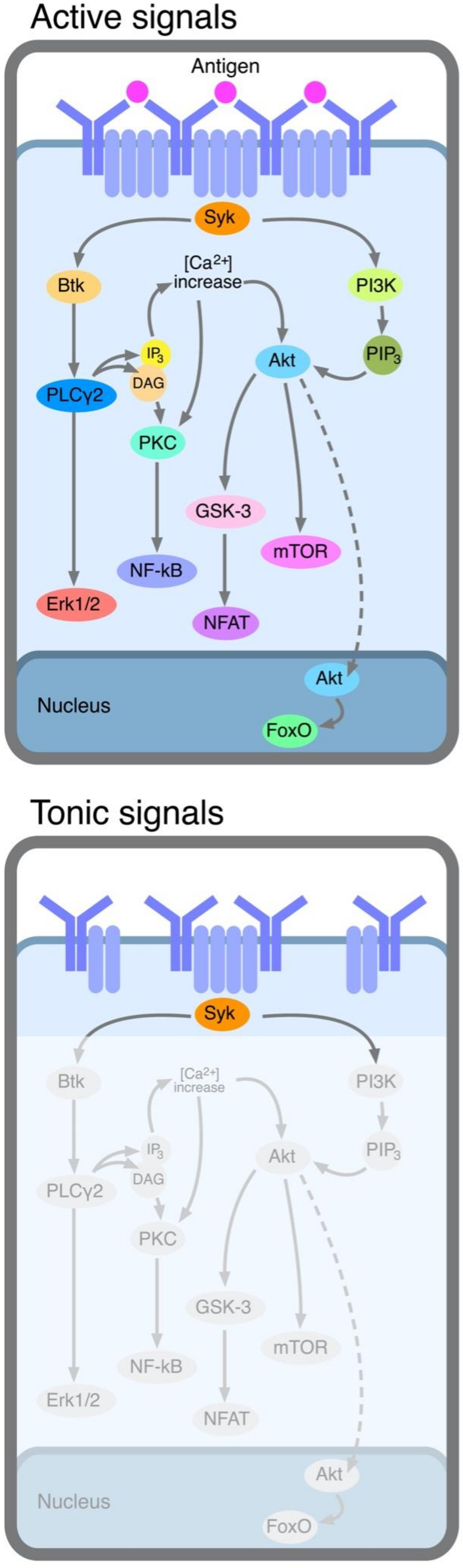
2.2. Active Signals
- (i)
-
Stimulation by self-antigens. The presence of frequent auto-immune manifestations, such as auto-immune hemolytic anemia or thrombocytopenia, suggested a connection between CLL and auto-immunity since the early studies [16]. This notion was substantiated more recently by the observation that a considerable number of CLL clones expressed a BcR characterized by poly-reactivity, a definition indicating that each monoclonal antibody could react with low affinity with a variety of different (auto)antigens, including platelets, aggregated IgG, nuclear antigens, double-strand (ds) and single-strand (ss) DNA, insulin, etc. Antibodies with these features are found among the “natural antibodies”, a family of antibodies mostly of the IgM isotype, of all mammalian species, representing one of the first lines of defense against assaulting pathogens [17][18]. These concepts were further refined by showing that U-CLL clones produce polyreactive IG very frequently, whereas this occurs rarely for M-CLL clones [19]. However, autoantibodies from patients with auto-immune manifestations, observed in both U- and M-CLL patients, are not produced by leukemic cells, since they are of the IgG isotype, they utilize both K and lambda light chain types and they are polyclonal [20]. Therefore, auto-immunity is not directly caused by the leukemic clone, although models in which leukemia and autoimmunity are part of the same pathogenetic process can be proposed, as researchers shall discuss later. Rare patients with cold agglutinin disease or cryoglobulinemia and CLL represent a notable exception. In these conditions, leukemic cells produce monoclonal low-affinity auto-antibodies to red cells or to the Fc portion of IgG [20][21][22], a feature consistent with the limited capacity of CLL cells to mature into plasma-cell-secreting antibodies [23]. Additional information came from the observation that poly-reactive antibodies often recognize antigens at the surface of apoptotic cells. Although normally located intracellularly, certain self-antigens can be expressed at the cell surface, when apoptosis is activated, and can be modified by metabolic processes, such as oxidation associated with apoptosis [24][25][26]. One physiologic function of poly-reactive antibodies is the clearance of apoptotic cells [27]. The expression of a BcR with polyreactive features, capable of recognizing apoptosis-related antigens, may become instrumental in promoting CLL clonal expansion, particularly in the presence of abundant CLL cell apoptosis. A substantial proportion of IG molecules cloned from or secreted by CLL cells react with intracellular proteins such as vimentin, tubulin and filamin B exposed at the cell surface following the induction of apoptosis [26]. Additional proteins to which these IGs have reactivity are those with which the sera of systemic lupus erythematosus (SLE) patients have reactivity, including Sm, snRNPA, Ku and other molecules which are also recognized both in the native or oxidized form by IG from the sera of SLE patients [24]. IG molecules with these reactivities are produced predominantly by U-CLL cases and many have stereotypic features, particularly those encoded for by the VH1-69 genes [24][25][26]. However, the absence of IGHV mutations and/or the utilization of a given stereotype do not classify these BcRs as specific for apoptosis-related antigens, since several of them fail to show any reactivity and different reactivities have been detected for the different BcR or BcR families investigated.
- (ii)
-
Stimulation by microbial antigens. In the absence of a clinically evident infection, such as the HP infection in gastric lymphoma, it is difficult to determine whether the BcRs of CLL cells may have specificity for antigens of a given pathogen. Nevertheless, researchers were able to trace a BcR with specificity for certain microorganisms in CLL. Hoogeboon and colleagues [28] analyzed 82 CLL patients, whose cells expressed an IGHV3-7-encoded BcR. The choice of this cohort was suggested by the reported over-representation of this gene in CLL and by the observation of a frequent SHM in these CLL clones, indicating antigenic stimulation and passage through germinal centers. A further selection within the cohort led to the choice of four patients in whom the BcR was characterized by a very short HCDR3 sequence of 5–6 amino acids (aa) instead of the canonical 15 aa. These BcRs were characterized by the utilization of nearly identical IGKV2-24-encoded Ig light chains and for sharing a glutamic acid at position 106 of HCDR3, which was not detected in any of the other sequences utilizing the IGHV3-7 gene. Cloning of genes and the expression of fully assembled IgM molecules led to the observation that the antibody bound 4/33 commensal yeast species and presented a specific binding to the β-(1,6)-glucan of the yeast. The substitution of the glutamic acid at position 106 of the HCDR3 via site-directed mutagenesis caused inhibition of the high-affinity binding to glucan, as did the substitution of certain aa of the short HCDR3. Finally, in vitro exposure of CLL cells with this BcR to the β-(1,6)-glucan resulted in specific cell proliferation. Together, these observations suggest a process of antigen selection, which may be more frequent than possibly thought, particularly if the stimulating antigens are carried by commensal rather than pathogenic microorganisms. Autoreactivity, described in the preceding section, and reactivity with microbial antigens may represent two aspects of the same phenomenon. Antibodies reacting with molecules exposed on the surface of apoptotic cells and/or with molecules oxidized following apoptosis show cross-reactivity for several microbial components. Therefore, this cross-reaction, which can be demonstrated in vitro, may also operate in vivo in CLL patients and certain CLL BcRs can concomitantly recognize antigens of micro-organisms and self-antigens [26].
- (iii)
-
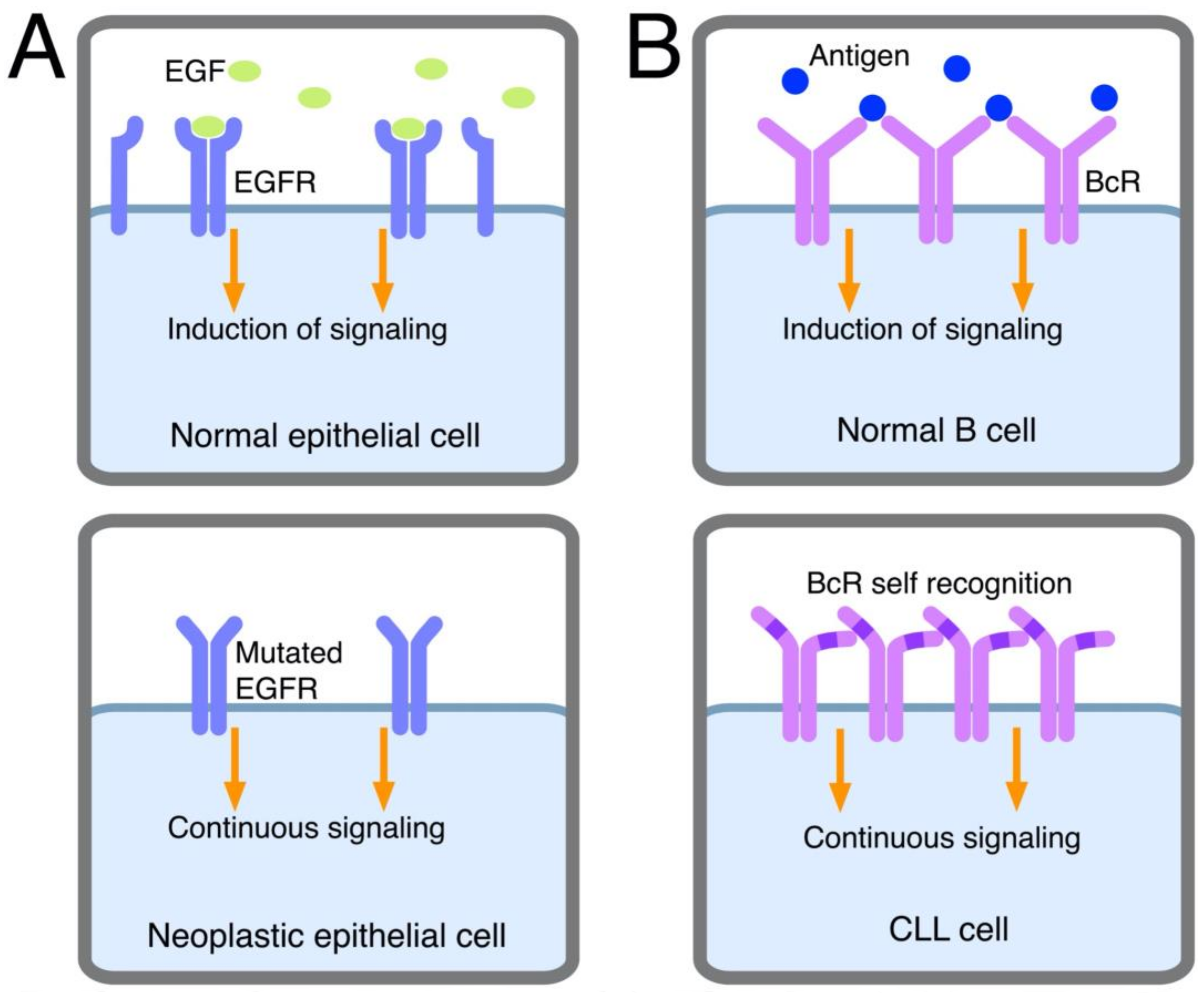
Autonomous signaling. This definition relates to the capacity of CLL cell BcRs to autonomously deliver activation signals to leukemic cells, as discovered in a very elegant system in vitro. Using retroviral gene transfer, BcRs from CLL clones were expressed in murine cells lacking endogenous BcR components and were thus unable to be signaled via their own BcR [29]. Positive signaling, revealed by Ca++ mobilization, was consistently noted following cell transfection with the BcRs from 17/17 CLL clones and not with the BcRs from other lymphoproliferative disorders including mantle cell, marginal zone and follicular lymphoma and myeloma (15/15 cases, collectively). This phenomenon was observed with the BcRs from U- and M-CLL cases and from cases expressing BcRs with/without poly-specific reactivity. Autonomous signaling also was observed with the BcRs from leukemic cell clones from mice that were transgenic for the T cell Leukemia 1 or tcl1 gene. These mice, obtained following the observation that U-CLL clones showed high TCL1 protein levels, expressed a tcl1 transgene under the control of the IGHV promotor and the Eμ-enhancer [30]. They developed a lymphoproliferative disorder characterized by the expression of CD5 and the utilization of unmutated IGHV genes, thus resembling human U-CLL [31]. Autonomous signaling was not observed when transfecting the BcRs from murine non-leukemic clones specific for several antigens, indicating that the phenomenon occurs preferentially in malignant cells [29]. Autonomous signaling can be reminiscent of that observed in other cancers, such as breast, lung or gastrointestinal tract cancers, in which activating mutations of receptors for growth factors enable the receptors to deliver signals similar to those of normal receptors following interaction with specific ligands [32][33]. Because of mutations, the receptor acquires the property of self-aggregation or dimerization or undergoes conformational changes, which can activate downstream signals (see Figure 2). However, there are no activating mutations in the BcR transducing the downstream signaling, and the system is more complex in comparison to these models. Ca++ mobilization is observed when the transfected BcR displays a HCDR3 motif which enables binding to a conserved epitope in the framework region 2 (FR2) of the IGHV domain of the same BcR molecule [29]. An alternative epitope for BcR autologous binding is found in FR3 [34]. Although these mechanisms have the same effect as the activating mutations of growth factor receptors present in other cancers, the underlying molecular mechanisms are different, as the recognition of a specific epitope of the “leukemic” BcR by the antigen-combining site of the same BcR expressed by the leukemic clone is required for receptor activation. In other words, autonomous signaling is intra-clonal and implies a specific interaction between BcR molecules on the surface of the same cell (or the BcRs of different cells from the same clone), given that the BcR recognizes a self-epitope (see Figure 2) [29]. A good example of the relevance of autonomous signaling is provided by a subgroup of CLL of the subset #2 stereotypes. These cases carry a single-point mutation, termed R110, at the junction between the variable and the constant region of the light chain. Although subset #2 stereotype is itself a poor prognosis indicator, R110+ cases display an even worse outcome. Light-chain mutation has been found to facilitate homotypic BcR-BcR interactions which result in a more robust activation of autonomous signaling [35].
3. Outcome of BcR Engagement
3.1. Identification of the Proliferating Cell Fraction of the CLL Clone
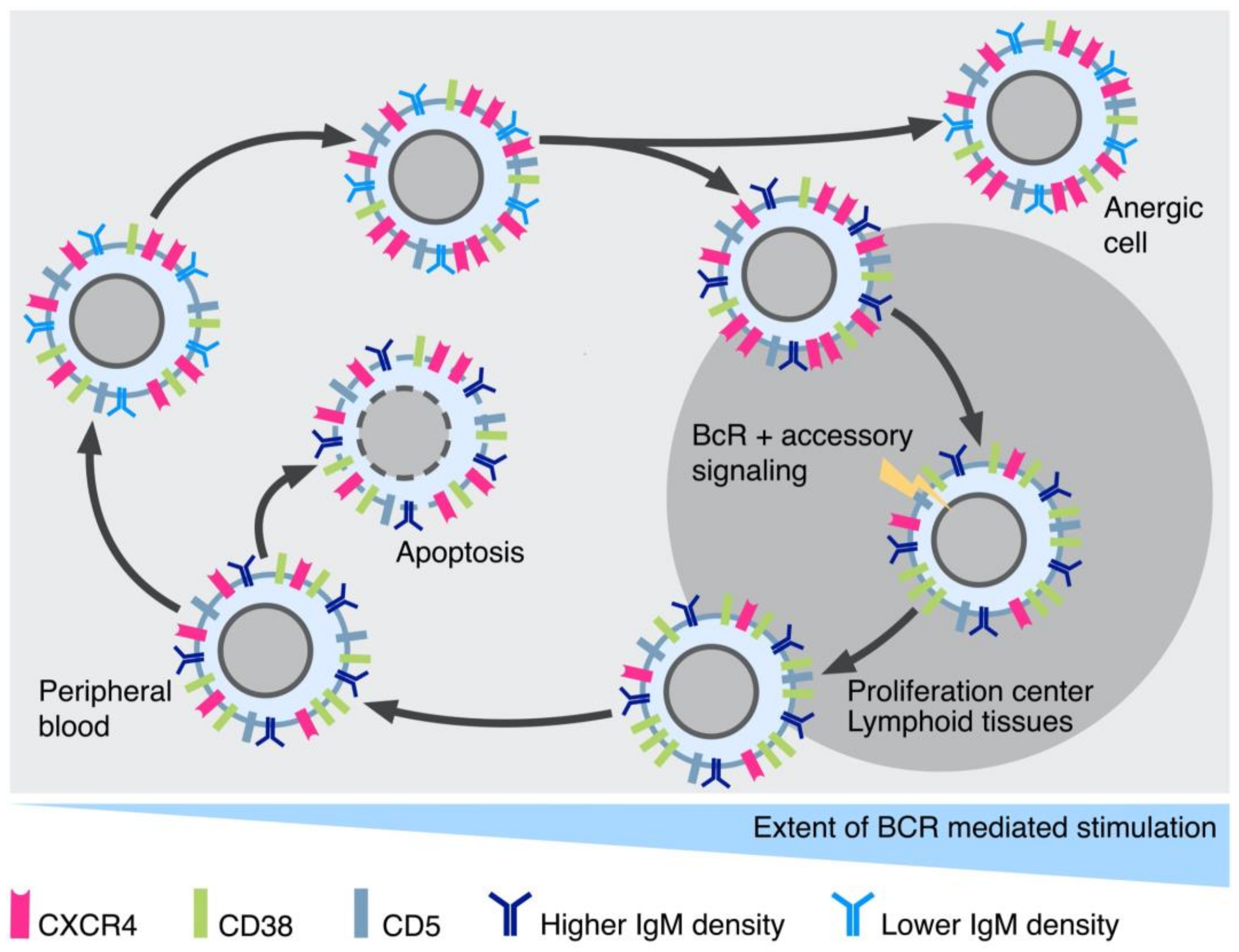
3.2. Choice between Proliferation and Apoptosis following BcR Stimulation
3.3. Tolerance of CLL Cells
3.4. The Function of BcR Other Than IgM
4. Inhibition of BcR-Dependent Tyrosine Kinases (TK)
References
- Clarke, M.F. Clinical and Therapeutic Implications of Cancer Stem Cells. N. Engl. J. Med. 2019, 380, 2237–2245.
- Isaacson, P.G.; Du, M.-Q. MALT Lymphoma: From Morphology to Molecules. Nat. Rev. Cancer 2004, 4, 644–653.
- Kuo, S.-H.; Yeh, K.-H.; Wu, M.-S.; Lin, C.-W.; Hsu, P.-N.; Wang, H.-P.; Chen, L.-T.; Cheng, A.-L. Helicobacter pylori Eradication Therapy Is Effective in the Treatment of Early-Stage H pylori-Positive Gastric Diffuse Large B-Cell Lymphomas. Blood 2012, 119, 4838–4844, quiz 5057.
- Quinn, E.R.; Chan, C.H.; Hadlock, K.G.; Foung, S.K.; Flint, M.; Levy, S. The B-Cell Receptor of a Hepatitis C Virus (HCV)-Associated Non-Hodgkin Lymphoma Binds the Viral E2 Envelope Protein, Implicating HCV in Lymphomagenesis. Blood 2001, 98, 3745–3749.
- Hermine, O.; Lefrère, F.; Bronowicki, J.-P.; Mariette, X.; Jondeau, K.; Eclache-Saudreau, V.; Delmas, B.; Valensi, F.; Cacoub, P.; Brechot, C.; et al. Regression of Splenic Lymphoma with Villous Lymphocytes after Treatment of Hepatitis C Virus Infection. N. Engl. J. Med. 2002, 347, 89–94.
- Young, R.M.; Staudt, L.M. Targeting Pathological B Cell Receptor Signalling in Lymphoid Malignancies. Nat. Rev. Drug Discov. 2013, 12, 229–243.
- Lam, K.P.; Kühn, R.; Rajewsky, K. In Vivo Ablation of Surface Immunoglobulin on Mature B Cells by Inducible Gene Targeting Results in Rapid Cell Death. Cell 1997, 90, 1073–1083.
- Kraus, M.; Alimzhanov, M.B.; Rajewsky, N.; Rajewsky, K. Survival of Resting Mature B Lymphocytes Depends on BCR Signaling via the Igalpha/Beta Heterodimer. Cell 2004, 117, 787–800.
- Yang, J.; Reth, M. Oligomeric Organization of the B-Cell Antigen Receptor on Resting Cells. Nature 2010, 467, 465–469.
- Yang, J.; Reth, M. The Dissociation Activation Model of B Cell Antigen Receptor Triggering. FEBS Lett. 2010, 584, 4872–4877.
- Srinivasan, L.; Sasaki, Y.; Calado, D.P.; Zhang, B.; Paik, J.H.; DePinho, R.A.; Kutok, J.L.; Kearney, J.F.; Otipoby, K.L.; Rajewsky, K. PI3 Kinase Signals BCR-Dependent Mature B Cell Survival. Cell 2009, 139, 573–586.
- Fais, F.; Sellars, B.; Ghiotto, F.; Yan, X.J.; Dono, M.; Allen, S.L.; Budman, D.; Dittmar, K.; Kolitz, J.; Lichtman, S.M.; et al. Examples of in Vivo Isotype Class Switching in IgM+ Chronic Lymphocytic Leukemia B Cells. J. Clin. Investig. 1996, 98, 1659–1666.
- Dono, M.; Hashimoto, S.; Fais, F.; Trejo, V.; Allen, S.L.; Lichtman, S.M.; Schulman, P.; Vinciguerra, V.P.; Sellars, B.; Gregersen, P.K.; et al. Evidence for Progenitors of Chronic Lymphocytic Leukemia B Cells That Undergo Intraclonal Differentiation and Diversification. Blood 1996, 87, 1586–1594.
- Agathangelidis, A.; Chatzidimitriou, A.; Gemenetzi, K.; Giudicelli, V.; Karypidou, M.; Plevova, K.; Davis, Z.; Yan, X.-J.; Jeromin, S.; Schneider, C.; et al. Higher-Order Connections between Stereotyped Subsets: Implications for Improved Patient Classification in CLL. Blood 2021, 137, 1365–1376.
- Gomes de Castro, M.A.; Wildhagen, H.; Sograte-Idrissi, S.; Hitzing, C.; Binder, M.; Trepel, M.; Engels, N.; Opazo, F. Differential Organization of Tonic and Chronic B Cell Antigen Receptors in the Plasma Membrane. Nat. Commun. 2019, 10, 820.
- Dameshek, W.; Schwartz, R.S. Leukemia and Auto-Immunization—Some Possible Relationships. Blood 1959, 14, 1151–1158.
- Sthoeger, Z.M.; Wakai, M.; Tse, D.B.; Vinciguerra, V.P.; Allen, S.L.; Budman, D.R.; Lichtman, S.M.; Schulman, P.; Weiselberg, L.R.; Chiorazzi, N. Production of Autoantibodies by CD5-Expressing B Lymphocytes from Patients with Chronic Lymphocytic Leukemia. J. Exp. Med. 1989, 169, 255–268.
- Borche, L.; Lim, A.; Binet, J.L.; Dighiero, G. Evidence That Chronic Lymphocytic Leukemia B Lymphocytes Are Frequently Committed to Production of Natural Autoantibodies. Blood 1990, 76, 562–569.
- Hervé, M.; Xu, K.; Ng, Y.-S.; Wardemann, H.; Albesiano, E.; Messmer, B.T.; Chiorazzi, N.; Meffre, E. Unmutated and Mutated Chronic Lymphocytic Leukemias Derive from Self-Reactive B Cell Precursors despite Expressing Different Antibody Reactivity. J. Clin. Investig. 2005, 115, 1636–1643.
- Molica, S.; Polliack, A. Autoimmune Hemolytic Anemia (AIHA) Associated with Chronic Lymphocytic Leukemia in the Current Era of Targeted Therapy. Leuk. Res. 2016, 50, 31–36.
- Tucci, F.A.; Kitanovski, S.; Johansson, P.; Klein-Hitpass, L.; Kahraman, A.; Dürig, J.; Hoffmann, D.; Küppers, R. Biased IGH VDJ Gene Repertoire and Clonal Expansions in B Cells of Chronically Hepatitis C Virus-Infected Individuals. Blood 2018, 131, 546–557.
- Stevenson, F.K.; Smith, G.J.; North, J.; Hamblin, T.J.; Glennie, M.J. Identification of Normal B-Cell Counterparts of Neoplastic Cells Which Secrete Cold Agglutinins of Anti-I and Anti-i Specificity. Br. J. Haematol. 1989, 72, 9–15.
- Rubartelli, A.; Sitia, R.; Zicca, A.; Grossi, C.E.; Ferrarini, M. Differentiation of Chronic Lymphocytic Leukemia Cells: Correlation between the Synthesis and Secretion of Immunoglobulins and the Ultrastructure of the Malignant Cells. Blood 1983, 62, 495–504.
- Catera, R.; Silverman, G.J.; Hatzi, K.; Seiler, T.; Didier, S.; Zhang, L.; Hervé, M.; Meffre, E.; Oscier, D.G.; Vlassara, H.; et al. Chronic Lymphocytic Leukemia Cells Recognize Conserved Epitopes Associated with Apoptosis and Oxidation. Mol. Med. Camb. Mass 2008, 14, 665–674.
- Chu, C.C.; Catera, R.; Zhang, L.; Didier, S.; Agagnina, B.M.; Damle, R.N.; Kaufman, M.S.; Kolitz, J.E.; Allen, S.L.; Rai, K.R.; et al. Many Chronic Lymphocytic Leukemia Antibodies Recognize Apoptotic Cells with Exposed Nonmuscle Myosin Heavy Chain IIA: Implications for Patient Outcome and Cell of Origin. Blood 2010, 115, 3907–3915.
- Lanemo Myhrinder, A.; Hellqvist, E.; Sidorova, E.; Söderberg, A.; Baxendale, H.; Dahle, C.; Willander, K.; Tobin, G.; Bäckman, E.; Söderberg, O.; et al. A New Perspective: Molecular Motifs on Oxidized LDL, Apoptotic Cells, and Bacteria Are Targets for Chronic Lymphocytic Leukemia Antibodies. Blood 2008, 111, 3838–3848.
- Peng, Y.; Kowalewski, R.; Kim, S.; Elkon, K.B. The Role of IgM Antibodies in the Recognition and Clearance of Apoptotic Cells. Mol. Immunol. 2005, 42, 781–787.
- Hoogeboom, R.; van Kessel, K.P.M.; Hochstenbach, F.; Wormhoudt, T.A.; Reinten, R.J.A.; Wagner, K.; Kater, A.P.; Guikema, J.E.J.; Bende, R.J.; van Noesel, C.J.M. A Mutated B Cell Chronic Lymphocytic Leukemia Subset That Recognizes and Responds to Fungi. J. Exp. Med. 2013, 210, 59–70.
- Dühren-von Minden, M.; Übelhart, R.; Schneider, D.; Wossning, T.; Bach, M.P.; Buchner, M.; Hofmann, D.; Surova, E.; Follo, M.; Köhler, F.; et al. Chronic Lymphocytic Leukaemia Is Driven by Antigen-Independent Cell-Autonomous Signalling. Nature 2012, 489, 309–312.
- Bichi, R.; Shinton, S.A.; Martin, E.S.; Koval, A.; Calin, G.A.; Cesari, R.; Russo, G.; Hardy, R.R.; Croce, C.M. Human Chronic Lymphocytic Leukemia Modeled in Mouse by Targeted TCL1 Expression. Proc. Natl. Acad. Sci. USA 2002, 99, 6955–6960.
- Chen, S.-S.; Batliwalla, F.; Holodick, N.E.; Yan, X.-J.; Yancopoulos, S.; Croce, C.M.; Rothstein, T.L.; Chiorazzi, N. Autoantigen Can Promote Progression to a More Aggressive TCL1 Leukemia by Selecting Variants with Enhanced B-Cell Receptor Signaling. Proc. Natl. Acad. Sci. USA 2013, 110, E1500–E1507.
- Chong, C.R.; Jänne, P.A. The Quest to Overcome Resistance to EGFR-Targeted Therapies in Cancer. Nat. Med. 2013, 19, 1389–1400.
- Turner, N.; Grose, R. Fibroblast Growth Factor Signalling: From Development to Cancer. Nat. Rev. Cancer 2010, 10, 116–129.
- Binder, M.; Müller, F.; Frick, M.; Wehr, C.; Simon, F.; Leistler, B.; Veelken, H.; Mertelsmann, R.; Trepel, M. CLL B-Cell Receptors Can Recognize Themselves: Alternative Epitopes and Structural Clues for Autostimulatory Mechanisms in CLL. Blood 2013, 121, 239–241.
- Maity, P.C.; Bilal, M.; Koning, M.T.; Young, M.; van Bergen, C.A.M.; Renna, V.; Nicolò, A.; Datta, M.; Gentner-Göbel, E.; Barendse, R.S.; et al. IGLV3-21*01 Is an Inherited Risk Factor for CLL through the Acquisition of a Single-Point Mutation Enabling Autonomous BCR Signaling. Proc. Natl. Acad. Sci. USA 2020, 117, 4320–4327.
- Ponzoni, M.; Doglioni, C.; Caligaris-Cappio, F. Chronic Lymphocytic Leukemia: The Pathologist’s View of Lymph Node Microenvironment. Semin. Diagn. Pathol. 2011, 28, 161–166.
- Morabito, F.; Shanafelt, T.D.; Gentile, M.; Reda, G.; Mauro, F.R.; Rossi, D.; Di Renzo, N.; Molica, S.; Angrilli, F.; Chiarenza, A.; et al. Immunoglobulin Heavy Chain Variable Region Gene and Prediction of Time to First Treatment in Patients with Chronic Lymphocytic Leukemia: Mutational Load or Mutational Status? Analysis of 1003 Cases. Am. J. Hematol. 2018, 93, E216–E219.
- Kaufman, M.; Yan, X.-J.; Li, W.; Ghia, E.M.; Langerak, A.W.; Rassenti, L.Z.; Belessi, C.; Kay, N.E.; Davi, F.; Byrd, J.C.; et al. Impact of the Types and Relative Quantities of IGHV Gene Mutations in Predicting Prognosis of Patients with Chronic Lymphocytic Leukemia. Front. Oncol. 2022, 12, 897280.
- Minici, C.; Gounari, M.; Übelhart, R.; Scarfò, L.; Dühren-von Minden, M.; Schneider, D.; Tasdogan, A.; Alkhatib, A.; Agathangelidis, A.; Ntoufa, S.; et al. Distinct Homotypic B-Cell Receptor Interactions Shape the Outcome of Chronic Lymphocytic Leukaemia. Nat. Commun. 2017, 8, 15746.
- Damle, R.N.; Ghiotto, F.; Valetto, A.; Albesiano, E.; Fais, F.; Yan, X.-J.; Sison, C.P.; Allen, S.L.; Kolitz, J.; Schulman, P.; et al. B-Cell Chronic Lymphocytic Leukemia Cells Express a Surface Membrane Phenotype of Activated, Antigen-Experienced B Lymphocytes. Blood 2002, 99, 4087–4093.
- Burger, J.A.; Chiorazzi, N. B Cell Receptor Signaling in Chronic Lymphocytic Leukemia. Trends Immunol. 2013, 34, 592–601.
- Zupo, S.; Isnardi, L.; Megna, M.; Massara, R.; Malavasi, F.; Dono, M.; Cosulich, E.; Ferrarini, M. CD38 Expression Distinguishes Two Groups of B-Cell Chronic Lymphocytic Leukemias with Different Responses to Anti-IgM Antibodies and Propensity to Apoptosis. Blood 1996, 88, 1365–1374.
- Malavasi, F.; Deaglio, S.; Damle, R.; Cutrona, G.; Ferrarini, M.; Chiorazzi, N. CD38 and Chronic Lymphocytic Leukemia: A Decade Later. Blood 2011, 118, 3470–3478.
- Chen, L.; Widhopf, G.; Huynh, L.; Rassenti, L.; Rai, K.R.; Weiss, A.; Kipps, T.J. Expression of ZAP-70 Is Associated with Increased B-Cell Receptor Signaling in Chronic Lymphocytic Leukemia. Blood 2002, 100, 4609–4614.
- Lanham, S.; Hamblin, T.; Oscier, D.; Ibbotson, R.; Stevenson, F.; Packham, G. Differential Signaling via Surface IgM Is Associated with VH Gene Mutational Status and CD38 Expression in Chronic Lymphocytic Leukemia. Blood 2003, 101, 1087–1093.
- Cutrona, G.; Colombo, M.; Matis, S.; Fabbi, M.; Spriano, M.; Callea, V.; Vigna, E.; Gentile, M.; Zupo, S.; Chiorazzi, N.; et al. Clonal Heterogeneity in Chronic Lymphocytic Leukemia Cells: Superior Response to Surface IgM Cross-Linking in CD38, ZAP-70-Positive Cells. Haematologica 2008, 93, 413–422.
- Damle, R.N.; Temburni, S.; Calissano, C.; Yancopoulos, S.; Banapour, T.; Sison, C.; Allen, S.L.; Rai, K.R.; Chiorazzi, N. CD38 Expression Labels an Activated Subset within Chronic Lymphocytic Leukemia Clones Enriched in Proliferating B Cells. Blood 2007, 110, 3352–3359.
- Mazzarello, A.N.; Fitch, M.; Hellerstein, M.K.; Chiorazzi, N. Measurement of Leukemic B-Cell Growth Kinetics in Patients with Chronic Lymphocytic Leukemia. Methods Mol. Biol. Clifton N. J. 2019, 1881, 129–151.
- Calissano, C.; Damle, R.N.; Hayes, G.; Murphy, E.J.; Hellerstein, M.K.; Moreno, C.; Sison, C.; Kaufman, M.S.; Kolitz, J.E.; Allen, S.L.; et al. In Vivo Intraclonal and Interclonal Kinetic Heterogeneity in B-Cell Chronic Lymphocytic Leukemia. Blood 2009, 114, 4832–4842.
- Herndon, T.M.; Chen, S.-S.; Saba, N.S.; Valdez, J.; Emson, C.; Gatmaitan, M.; Tian, X.; Hughes, T.E.; Sun, C.; Arthur, D.C.; et al. Direct in Vivo Evidence for Increased Proliferation of CLL Cells in Lymph Nodes Compared to Bone Marrow and Peripheral Blood. Leukemia 2017, 31, 1340–1347.
- Herishanu, Y.; Pérez-Galán, P.; Liu, D.; Biancotto, A.; Pittaluga, S.; Vire, B.; Gibellini, F.; Njuguna, N.; Lee, E.; Stennett, L.; et al. The Lymph Node Microenvironment Promotes B-Cell Receptor Signaling, NF-KappaB Activation, and Tumor Proliferation in Chronic Lymphocytic Leukemia. Blood 2011, 117, 563–574.
- Ferrer, G.; Jung, B.; Chiu, P.Y.; Aslam, R.; Palacios, F.; Mazzarello, A.N.; Vergani, S.; Bagnara, D.; Chen, S.-S.; Yancopoulos, S.; et al. Myeloid-Derived Suppressor Cell Subtypes Differentially Influence T-Cell Function, T-Helper Subset Differentiation, and Clinical Course in CLL. Leukemia 2021, 35, 3163–3175.
- Burger, J.A.; Kipps, T.J. CXCR4: A Key Receptor in the Crosstalk between Tumor Cells and Their Microenvironment. Blood 2006, 107, 1761–1767.
- Burger, J.A.; Quiroga, M.P.; Hartmann, E.; Bürkle, A.; Wierda, W.G.; Keating, M.J.; Rosenwald, A. High-Level Expression of the T-Cell Chemokines CCL3 and CCL4 by Chronic Lymphocytic Leukemia B Cells in Nurselike Cell Cocultures and after BCR Stimulation. Blood 2009, 113, 3050–3058.
- Messmer, D.; Fecteau, J.-F.; O’Hayre, M.; Bharati, I.S.; Handel, T.M.; Kipps, T.J. Chronic Lymphocytic Leukemia Cells Receive RAF-Dependent Survival Signals in Response to CXCL12 That Are Sensitive to Inhibition by Sorafenib. Blood 2011, 117, 882–889.
- Vlad, A.; Deglesne, P.-A.; Letestu, R.; Saint-Georges, S.; Chevallier, N.; Baran-Marszak, F.; Varin-Blank, N.; Ajchenbaum-Cymbalista, F.; Ledoux, D. Down-Regulation of CXCR4 and CD62L in Chronic Lymphocytic Leukemia Cells Is Triggered by B-Cell Receptor Ligation and Associated with Progressive Disease. Cancer Res. 2009, 69, 6387–6395.
- Burgueño-Bucio, E.; Mier-Aguilar, C.A.; Soldevila, G. The Multiple Faces of CD5. J. Leukoc. Biol. 2019, 105, 891–904.
- Calissano, C.; Damle, R.N.; Marsilio, S.; Yan, X.-J.; Yancopoulos, S.; Hayes, G.; Emson, C.; Murphy, E.J.; Hellerstein, M.K.; Sison, C.; et al. Intraclonal Complexity in Chronic Lymphocytic Leukemia: Fractions Enriched in Recently Born/Divided and Older/Quiescent Cells. Mol. Med. Camb. Mass 2011, 17, 1374–1382.
- Bartholdy, B.A.; Wang, X.; Yan, X.-J.; Pascual, M.; Fan, M.; Barrientos, J.; Allen, S.L.; Martinez-Climent, J.A.; Rai, K.R.; Chiorazzi, N.; et al. CLL Intraclonal Fractions Exhibit Established and Recently Acquired Patterns of DNA Methylation. Blood Adv. 2020, 4, 893–905.
- Cui, B.; Chen, L.; Zhang, S.; Mraz, M.; Fecteau, J.-F.; Yu, J.; Ghia, E.M.; Zhang, L.; Bao, L.; Rassenti, L.Z.; et al. MicroRNA-155 Influences B-Cell Receptor Signaling and Associates with Aggressive Disease in Chronic Lymphocytic Leukemia. Blood 2014, 124, 546–554.
- D’Avola, A.; Drennan, S.; Tracy, I.; Henderson, I.; Chiecchio, L.; Larrayoz, M.; Rose-Zerilli, M.; Strefford, J.; Plass, C.; Johnson, P.W.; et al. Surface IgM Expression and Function Are Associated with Clinical Behavior, Genetic Abnormalities, and DNA Methylation in CLL. Blood 2016, 128, 816–826.
- Efremov, D.G.; Gobessi, S.; Longo, P.G. Signaling Pathways Activated by Antigen-Receptor Engagement in Chronic Lymphocytic Leukemia B-Cells. Autoimmun. Rev. 2007, 7, 102–108.
- Guarini, A.; Chiaretti, S.; Tavolaro, S.; Maggio, R.; Peragine, N.; Citarella, F.; Ricciardi, M.R.; Santangelo, S.; Marinelli, M.; De Propris, M.S.; et al. BCR Ligation Induced by IgM Stimulation Results in Gene Expression and Functional Changes Only in IgV H Unmutated Chronic Lymphocytic Leukemia (CLL) Cells. Blood 2008, 112, 782–792.
- Lagneaux, L.; Delforge, A.; Bron, D.; De Bruyn, C.; Stryckmans, P. Chronic Lymphocytic Leukemic B Cells but Not Normal B Cells Are Rescued from Apoptosis by Contact with Normal Bone Marrow Stromal Cells. Blood 1998, 91, 2387–2396.
- Burger, J.A.; Tsukada, N.; Burger, M.; Zvaifler, N.J.; Dell’Aquila, M.; Kipps, T.J. Blood-Derived Nurse-like Cells Protect Chronic Lymphocytic Leukemia B Cells from Spontaneous Apoptosis through Stromal Cell-Derived Factor-1. Blood 2000, 96, 2655–2663.
- Pedersen, I.M.; Kitada, S.; Leoni, L.M.; Zapata, J.M.; Karras, J.G.; Tsukada, N.; Kipps, T.J.; Choi, Y.S.; Bennett, F.; Reed, J.C. Protection of CLL B Cells by a Follicular Dendritic Cell Line Is Dependent on Induction of Mcl-1. Blood 2002, 100, 1795–1801.
- Zucchetto, A.; Benedetti, D.; Tripodo, C.; Bomben, R.; Dal Bo, M.; Marconi, D.; Bossi, F.; Lorenzon, D.; Degan, M.; Rossi, F.M.; et al. CD38/CD31, the CCL3 and CCL4 Chemokines, and CD49d/Vascular Cell Adhesion Molecule-1 Are Interchained by Sequential Events Sustaining Chronic Lymphocytic Leukemia Cell Survival. Cancer Res. 2009, 69, 4001–4009.
- Arruga, F.; Gyau, B.B.; Iannello, A.; Vitale, N.; Vaisitti, T.; Deaglio, S. Immune Response Dysfunction in Chronic Lymphocytic Leukemia: Dissecting Molecular Mechanisms and Microenvironmental Conditions. Int. J. Mol. Sci. 2020, 21, 1825.
- Fabbri, G.; Dalla-Favera, R. The Molecular Pathogenesis of Chronic Lymphocytic Leukaemia. Nat. Rev. Cancer 2016, 16, 145–162.
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent Deletions and Down-Regulation of Micro- RNA Genes MiR15 and MiR16 at 13q14 in Chronic Lymphocytic Leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529.
- Klein, U.; Lia, M.; Crespo, M.; Siegel, R.; Shen, Q.; Mo, T.; Ambesi-Impiombato, A.; Califano, A.; Migliazza, A.; Bhagat, G.; et al. The DLEU2/MiR-15a/16-1 Cluster Controls B Cell Proliferation and Its Deletion Leads to Chronic Lymphocytic Leukemia. Cancer Cell 2010, 17, 28–40.
- Cimmino, A.; Calin, G.A.; Fabbri, M.; Iorio, M.V.; Ferracin, M.; Shimizu, M.; Wojcik, S.E.; Aqeilan, R.I.; Zupo, S.; Dono, M.; et al. MiR-15 and MiR-16 Induce Apoptosis by Targeting BCL2. Proc. Natl. Acad. Sci. USA 2005, 102, 13944–13949.
- Cutrona, G.; Matis, S.; Colombo, M.; Massucco, C.; Baio, G.; Valdora, F.; Emionite, L.; Fabris, S.; Recchia, A.G.; Gentile, M.; et al. Effects of MiRNA-15 and MiRNA-16 Expression Replacement in Chronic Lymphocytic Leukemia: Implication for Therapy. Leukemia 2017, 31, 1894–1904.
- Roberts, A.W. Therapeutic Development and Current Uses of BCL-2 Inhibition. Hematol. Am. Soc. Hematol. Educ. Program 2020, 2020, 1–9.
- Fischer, K.; Al-Sawaf, O.; Bahlo, J.; Fink, A.-M.; Tandon, M.; Dixon, M.; Robrecht, S.; Warburton, S.; Humphrey, K.; Samoylova, O.; et al. Venetoclax and Obinutuzumab in Patients with CLL and Coexisting Conditions. N. Engl. J. Med. 2019, 380, 2225–2236.
- Bruno, S.; Ghiotto, F.; Tenca, C.; Mazzarello, A.N.; Bono, M.; Luzzi, P.; Casciaro, S.; Recchia, A.; DeCensi, A.; Morabito, F.; et al. N-(4-Hydroxyphenyl)Retinamide Promotes Apoptosis of Resting and Proliferating B-Cell Chronic Lymphocytic Leukemia Cells and Potentiates Fludarabine and ABT-737 Cytotoxicity. Leukemia 2012, 26, 2260–2268.
- Ghiotto, F.; Fais, F.; Bruno, S. BH3-Only Proteins: The Death-Puppeteer’s Wires. Cytom. Part J. Int. Soc. Anal. Cytol. 2010, 77, 11–21.
- Yeomans, A.; Thirdborough, S.M.; Valle-Argos, B.; Linley, A.; Krysov, S.; Hidalgo, M.S.; Leonard, E.; Ishfaq, M.; Wagner, S.D.; Willis, A.E.; et al. Engagement of the B-Cell Receptor of Chronic Lymphocytic Leukemia Cells Drives Global and MYC-Specific MRNA Translation. Blood 2016, 127, 449–457.
- Öztürk, S.; Paul, Y.; Afzal, S.; Gil-Farina, I.; Jauch, A.; Bruch, P.-M.; Kalter, V.; Hanna, B.; Arseni, L.; Roessner, P.M.; et al. Longitudinal Analyses of CLL in Mice Identify Leukemia-Related Clonal Changes Including a Myc Gain Predicting Poor Outcome in Patients. Leukemia 2022, 36, 464–475.
- Biran, A.; Yin, S.; Kretzmer, H.; Ten Hacken, E.; Parvin, S.; Lucas, F.; Uduman, M.; Gutierrez, C.; Dangle, N.; Billington, L.; et al. Activation of Notch and Myc Signaling via B-Cell-Restricted Depletion of Dnmt3a Generates a Consistent Murine Model of Chronic Lymphocytic Leukemia. Cancer Res. 2021, 81, 6117–6130.
- Hayakawa, K.; Formica, A.M.; Brill-Dashoff, J.; Shinton, S.A.; Ichikawa, D.; Zhou, Y.; Morse, H.C.; Hardy, R.R. Early Generated B1 B Cells with Restricted BCRs Become Chronic Lymphocytic Leukemia with Continued C-Myc and Low Bmf Expression. J. Exp. Med. 2016, 213, 3007–3024.
- Evan, G.I.; Wyllie, A.H.; Gilbert, C.S.; Littlewood, T.D.; Land, H.; Brooks, M.; Waters, C.M.; Penn, L.Z.; Hancock, D.C. Induction of Apoptosis in Fibroblasts by C-Myc Protein. Cell 1992, 69, 119–128.
- Cutrona, G.; Ulivi, M.; Fais, F.; Roncella, S.; Ferrarini, M. Transfection of the C-Myc Oncogene into Normal Epstein-Barr Virus-Harboring B Cells Results in New Phenotypic and Functional Features Resembling Those of Burkitt Lymphoma Cells and Normal Centroblasts. J. Exp. Med. 1995, 181, 699–711.
- Cutrona, G.; Dono, M.; Pastorino, S.; Ulivi, M.; Burgio, V.L.; Zupo, S.; Roncella, S.; Ferrarini, M. The Propensity to Apoptosis of Centrocytes and Centroblasts Correlates with Elevated Levels of Intracellular Myc Protein. Eur. J. Immunol. 1997, 27, 234–238.
- Calado, D.P.; Sasaki, Y.; Godinho, S.A.; Pellerin, A.; Köchert, K.; Sleckman, B.P.; de Alborán, I.M.; Janz, M.; Rodig, S.; Rajewsky, K. The Cell-Cycle Regulator c-Myc Is Essential for the Formation and Maintenance of Germinal Centers. Nat. Immunol. 2012, 13, 1092–1100.
- Dominguez-Sola, D.; Victora, G.D.; Ying, C.Y.; Phan, R.T.; Saito, M.; Nussenzweig, M.C.; Dalla-Favera, R. The Proto-Oncogene MYC Is Required for Selection in the Germinal Center and Cyclic Reentry. Nat. Immunol. 2012, 13, 1083–1091.
- Kluckova, K.; Clear, A.J.; D’Avola, A.; Rassenti, L.Z.; Kipps, T.J.; Gribben, J.G.; Riches, J.C. B-Cell Receptor Signaling Induced Metabolic Alterations in Chronic Lymphocytic Leukemia Can Be Partially Bypassed by TP53 Abnormalities. HemaSphere 2022, 6, e722.
- Linley, A.; Valle-Argos, B.; Steele, A.J.; Stevenson, F.K.; Forconi, F.; Packham, G. Higher Levels of Reactive Oxygen Species Are Associated with Anergy in Chronic Lymphocytic Leukemia. Haematologica 2015, 100, e265–e268.
- Hondares, E.; Brown, M.A.; Musset, B.; Morgan, D.; Cherny, V.V.; Taubert, C.; Bhamrah, M.K.; Coe, D.; Marelli-Berg, F.; Gribben, J.G.; et al. Enhanced Activation of an Amino-Terminally Truncated Isoform of the Voltage-Gated Proton Channel HVCN1 Enriched in Malignant B Cells. Proc. Natl. Acad. Sci. USA 2014, 111, 18078–18083.
- Muzio, M.; Apollonio, B.; Scielzo, C.; Frenquelli, M.; Vandoni, I.; Boussiotis, V.; Caligaris-Cappio, F.; Ghia, P. Constitutive Activation of Distinct BCR-Signaling Pathways in a Subset of CLL Patients: A Molecular Signature of Anergy. Blood 2008, 112, 188–195.
- Mockridge, C.I.; Potter, K.N.; Wheatley, I.; Neville, L.A.; Packham, G.; Stevenson, F.K. Reversible Anergy of SIgM-Mediated Signaling in the Two Subsets of CLL Defined by VH-Gene Mutational Status. Blood 2007, 109, 4424–4431.
- Apollonio, B.; Scielzo, C.; Bertilaccio, M.T.S.; Ten Hacken, E.; Scarfò, L.; Ranghetti, P.; Stevenson, F.; Packham, G.; Ghia, P.; Muzio, M.; et al. Targeting B-Cell Anergy in Chronic Lymphocytic Leukemia. Blood 2013, 121, 3879–3888.
- Tibaldi, E.; Brunati, A.M.; Zonta, F.; Frezzato, F.; Gattazzo, C.; Zambello, R.; Gringeri, E.; Semenzato, G.; Pagano, M.A.; Trentin, L. Lyn-Mediated SHP-1 Recruitment to CD5 Contributes to Resistance to Apoptosis of B-Cell Chronic Lymphocytic Leukemia Cells. Leukemia 2011, 25, 1768–1781.
- Goodnow, C.C.; Sprent, J.; Fazekas de St Groth, B.; Vinuesa, C.G. Cellular and Genetic Mechanisms of Self Tolerance and Autoimmunity. Nature 2005, 435, 590–597.
- Cashman, K.S.; Jenks, S.A.; Woodruff, M.C.; Tomar, D.; Tipton, C.M.; Scharer, C.D.; Eun-Hyung Lee, F.; Boss, J.M.; Sanz, I. Understanding and Measuring Human B-Cell Tolerance and Its Breakdown in Autoimmune Disease. Immunol. Rev. 2019, 292, 76–89.
- Duty, J.A.; Szodoray, P.; Zheng, N.-Y.; Koelsch, K.A.; Zhang, Q.; Swiatkowski, M.; Mathias, M.; Garman, L.; Helms, C.; Nakken, B.; et al. Functional Anergy in a Subpopulation of Naive B Cells from Healthy Humans That Express Autoreactive Immunoglobulin Receptors. J. Exp. Med. 2009, 206, 139–151.
- Agathangelidis, A.; Darzentas, N.; Hadzidimitriou, A.; Brochet, X.; Murray, F.; Yan, X.-J.; Davis, Z.; van Gastel-Mol, E.J.; Tresoldi, C.; Chu, C.C.; et al. Stereotyped B-Cell Receptors in One-Third of Chronic Lymphocytic Leukemia: A Molecular Classification with Implications for Targeted Therapies. Blood 2012, 119, 4467–4475.
- Wan, Z.; Zhao, Y.; Sun, Y. Immunoglobulin D and Its Encoding Genes: An Updated Review. Dev. Comp. Immunol. 2021, 124, 104198.
- Übelhart, R.; Hug, E.; Bach, M.P.; Wossning, T.; Dühren-von Minden, M.; Horn, A.H.C.; Tsiantoulas, D.; Kometani, K.; Kurosaki, T.; Binder, C.J.; et al. Responsiveness of B Cells Is Regulated by the Hinge Region of IgD. Nat. Immunol. 2015, 16, 534–543.
- Murphy, E.J.; Neuberg, D.S.; Rassenti, L.Z.; Hayes, G.; Redd, R.; Emson, C.; Li, K.; Brown, J.R.; Wierda, W.G.; Turner, S.; et al. Leukemia-Cell Proliferation and Disease Progression in Patients with Early Stage Chronic Lymphocytic Leukemia. Leukemia 2017, 31, 1348–1354.
- Mazzarello, A.N.; Gentner-Göbel, E.; Dühren-von Minden, M.; Tarasenko, T.N.; Nicolò, A.; Ferrer, G.; Vergani, S.; Liu, Y.; Bagnara, D.; Rai, K.R.; et al. B Cell Receptor Isotypes Differentially Associate with Cell Signaling, Kinetics, and Outcome in Chronic Lymphocytic Leukemia. J. Clin. Investig. 2022, 132, e149308.
- Maity, P.C.; Blount, A.; Jumaa, H.; Ronneberger, O.; Lillemeier, B.F.; Reth, M. B Cell Antigen Receptors of the IgM and IgD Classes Are Clustered in Different Protein Islands That Are Altered during B Cell Activation. Sci. Signal. 2015, 8, ra93.
- Kläsener, K.; Maity, P.C.; Hobeika, E.; Yang, J.; Reth, M. B Cell Activation Involves Nanoscale Receptor Reorganizations and Inside-out Signaling by Syk. eLife 2014, 3, e02069.
- Ten Hacken, E.; Sivina, M.; Kim, E.; O’Brien, S.; Wierda, W.G.; Ferrajoli, A.; Estrov, Z.; Keating, M.J.; Oellerich, T.; Scielzo, C.; et al. Functional Differences between IgM and IgD Signaling in Chronic Lymphocytic Leukemia. J. Immunol. Baltim. 2016, 197, 2522–2531.
- Haerzschel, A.; Catusse, J.; Hutterer, E.; Paunovic, M.; Zirlik, K.; Eibel, H.; Krenn, P.W.; Hartmann, T.N.; Burger, M. BCR and Chemokine Responses upon Anti-IgM and Anti-IgD Stimulation in Chronic Lymphocytic Leukaemia. Ann. Hematol. 2016, 95, 1979–1988.
- Zupo, S.; Massara, R.; Dono, M.; Rossi, E.; Malavasi, F.; Cosulich, M.E.; Ferrarini, M. Apoptosis or Plasma Cell Differentiation of CD38-Positive B-Chronic Lymphocytic Leukemia Cells Induced by Cross-Linking of Surface IgM or IgD. Blood 2000, 95, 1199–1206.
- Sabouri, Z.; Perotti, S.; Spierings, E.; Humburg, P.; Yabas, M.; Bergmann, H.; Horikawa, K.; Roots, C.; Lambe, S.; Young, C.; et al. IgD Attenuates the IgM-Induced Anergy Response in Transitional and Mature B Cells. Nat. Commun. 2016, 7, 13381.
- Vire, B.; David, A.; Wiestner, A. TOSO, the Fcmicro Receptor, Is Highly Expressed on Chronic Lymphocytic Leukemia B Cells, Internalizes upon IgM Binding, Shuttles to the Lysosome, and Is Downregulated in Response to TLR Activation. J. Immunol. Baltim. 2011, 187, 4040–4050.
- Kubagawa, H.; Oka, S.; Kubagawa, Y.; Torii, I.; Takayama, E.; Kang, D.-W.; Gartland, G.L.; Bertoli, L.F.; Mori, H.; Takatsu, H.; et al. Identity of the Elusive IgM Fc Receptor (FcmuR) in Humans. J. Exp. Med. 2009, 206, 2779–2793.
- Colombo, M.; Cutrona, G.; Reverberi, D.; Fabris, S.; Neri, A.; Fabbi, M.; Quintana, G.; Quarta, G.; Ghiotto, F.; Fais, F.; et al. Intraclonal Cell Expansion and Selection Driven by B Cell Receptor in Chronic Lymphocytic Leukemia. Mol. Med. Camb. Mass 2011, 17, 834–839.
- Burger, J.A. Treatment of Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2020, 383, 460–473.
- Burger, J.A.; Wiestner, A. Targeting B Cell Receptor Signalling in Cancer: Preclinical and Clinical Advances. Nat. Rev. Cancer 2018, 18, 148–167.
- Hallek, M.; Shanafelt, T.D.; Eichhorst, B. Chronic Lymphocytic Leukaemia. Lancet Lond. Engl. 2018, 391, 1524–1537.
- Stephens, D.M.; Byrd, J.C. How I Manage Ibrutinib Intolerance and Complications in Patients with Chronic Lymphocytic Leukemia. Blood 2019, 133, 1298–1307.
- Sharma, S.; Rai, K.R. Chronic Lymphocytic Leukemia (CLL) Treatment: So Many Choices, Such Great Options. Cancer 2019, 125, 1432–1440.
- Byrd, J.C.; Furman, R.R.; Coutre, S.E.; Flinn, I.W.; Burger, J.A.; Blum, K.A.; Grant, B.; Sharman, J.P.; Coleman, M.; Wierda, W.G.; et al. Targeting BTK with Ibrutinib in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2013, 369, 32–42.
- Woyach, J.A.; Smucker, K.; Smith, L.L.; Lozanski, A.; Zhong, Y.; Ruppert, A.S.; Lucas, D.; Williams, K.; Zhao, W.; Rassenti, L.; et al. Prolonged Lymphocytosis during Ibrutinib Therapy Is Associated with Distinct Molecular Characteristics and Does Not Indicate a Suboptimal Response to Therapy. Blood 2014, 123, 1810–1817.
- O’Brien, S.; Furman, R.R.; Coutre, S.; Flinn, I.W.; Burger, J.A.; Blum, K.; Sharman, J.; Wierda, W.; Jones, J.; Zhao, W.; et al. Single-Agent Ibrutinib in Treatment-Naïve and Relapsed/Refractory Chronic Lymphocytic Leukemia: A 5-Year Experience. Blood 2018, 131, 1910–1919.
- Ghia, P.; Pluta, A.; Wach, M.; Lysak, D.; Kozak, T.; Simkovic, M.; Kaplan, P.; Kraychok, I.; Illes, A.; de la Serna, J.; et al. ASCEND: Phase III, Randomized Trial of Acalabrutinib Versus Idelalisib Plus Rituximab or Bendamustine Plus Rituximab in Relapsed or Refractory Chronic Lymphocytic Leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 2849–2861.
- Beckmann, L.; Berg, V.; Dickhut, C.; Sun, C.; Merkel, O.; Bloehdorn, J.; Robrecht, S.; Seifert, M.; da Palma Guerreiro, A.; Claasen, J.; et al. MARCKS Affects Cell Motility and Response to BTK Inhibitors in CLL. Blood 2021, 138, 544–556.
- Drennan, S.; Chiodin, G.; D’Avola, A.; Tracy, I.; Johnson, P.W.; Trentin, L.; Steele, A.J.; Packham, G.; Stevenson, F.K.; Forconi, F. Ibrutinib Therapy Releases Leukemic Surface IgM from Antigen Drive in Chronic Lymphocytic Leukemia Patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 2503–2512.
- Burger, J.A. CLL Cells Are Moved by the MARCKS Brothers. Blood 2021, 138, 503–504.
- Pavlasova, G.; Borsky, M.; Seda, V.; Cerna, K.; Osickova, J.; Doubek, M.; Mayer, J.; Calogero, R.; Trbusek, M.; Pospisilova, S.; et al. Ibrutinib Inhibits CD20 Upregulation on CLL B Cells Mediated by the CXCR4/SDF-1 Axis. Blood 2016, 128, 1609–1613.
- Burger, J.A.; Landau, D.A.; Taylor-Weiner, A.; Bozic, I.; Zhang, H.; Sarosiek, K.; Wang, L.; Stewart, C.; Fan, J.; Hoellenriegel, J.; et al. Clonal Evolution in Patients with Chronic Lymphocytic Leukaemia Developing Resistance to BTK Inhibition. Nat. Commun. 2016, 7, 11589.
- Liu, T.-M.; Woyach, J.A.; Zhong, Y.; Lozanski, A.; Lozanski, G.; Dong, S.; Strattan, E.; Lehman, A.; Zhang, X.; Jones, J.A.; et al. Hypermorphic Mutation of Phospholipase C, Γ2 Acquired in Ibrutinib-Resistant CLL Confers BTK Independency upon B-Cell Receptor Activation. Blood 2015, 126, 61–68.


