 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Giancarlo Castaman | -- | 3209 | 2022-11-29 11:38:20 | | | |
| 2 | Lindsay Dong | Meta information modification | 3209 | 2022-11-29 12:41:43 | | |
Video Upload Options
The standard of care for hemophilia A has been intravenous administration of exogenous factor VIII (FVIII), either as prophylaxis or episodically. Gene therapy is a treatment strategy used to repair or provide a functional copy of a gene that is either absent or expressed as a non-functional protein. Treating a genetic disease with gene therapy requires that the transgene (or its protein product) be delivered to the physiologically relevant target tissue or tissues, be stably expressed, and not interfere with the functional integrity of the cells in these tissues. The ultimate goal of gene therapy for patients with hemophilia A is the production of a treatment that is given as a one-time infusion and that allows adequate long-term expression of the deficient FVIII, with the maintenance of steady-state plasma FVIII concentrations. This would minimize (or ideally eliminate) bleeding episodes and thereby decrease the patient and societal burden of the disease.
1. Introduction
2. Gene Therapy in Hemophilia A
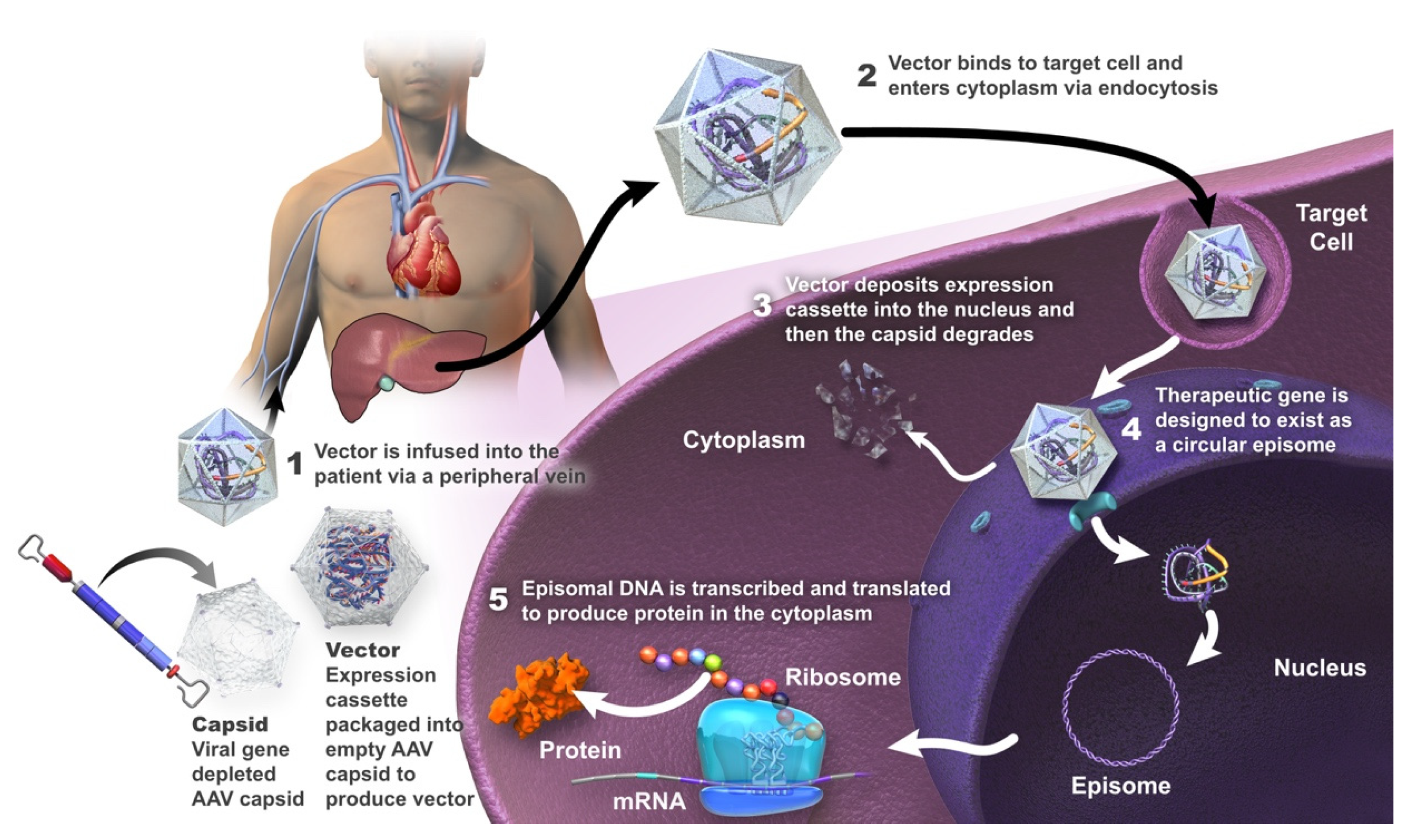
2.1. Valoctocogene Roxaparvovec
2.2. Dirloctocogene Samoparvovec
2.3. SPK-8016
2.4. Giroctocogene Fitelparvovec
2.5. TAK-754
2.6. BAY 2599023
2.7. AAV8-HLP-hFVIII-V3
3. Opportunities and Challenges of Gene Therapy in Hemophilia A
3.1. Opportunities
Gene therapy is a disease-transforming therapy that has the potential to become the new standard for the treatment of patients with hemophilia A [44]. It may enable the patient to achieve a sustained physiological level of endogenously produced FVIII protein that could provide effective prophylaxis without the need for exogenous factor replacement therapy [16][18]. Such continuous endogenous expression of physiological levels of FVIII can be expected to eliminate breakthrough bleeding and micro-hemorrhages [44]. Gene therapy may, thus, provide protection against further bleeding-induced joint damage [45]. Given that even minimal increases in FVIII levels can lead to significantly improved bleeding outcomes, it is expected that gene therapy will provide a resultant improvement in the QoL of the individual with hemophilia A [14][44].
3.2. Challenges
Successful gene therapy was first reported, in 2011, with intravenously administered AAV-based liver-directed gene therapy in patients with severe hemophilia B [46]. In contrast, gene therapy directed at hemophilia A has lagged behind that directed at hemophilia B, largely due to the limitations inherent in the size of the FVIII gene and the structure of FVIII, problems achieving therapeutic levels of the transgenic protein, and cellular immune responses to the capsid of the AAV vector [47] (also identified in trials of therapy for hemophilia B, although to a lesser extent), as well as issues related to the duration and variability of this form of therapy [1][4][6].
FVIII Structure
FVIII Expression
Infusion-Related Adverse Reactions
Anti-AAV Neutralizing Antibodies
Elevation of Liver Enzymes
Oncogenesis
Durability and Variability of Transgene Expression
Patient Expectations
4. Conclusions
References
- Arruda, V.R.; Weber, J.; Samelson-Jones, B.J. Gene Therapy for Inherited Bleeding Disorders. Semin. Thromb. Hemost. 2021, 47, 161–173.
- Berntorp, E.; Fischer, K.; Hart, D.P.; Mancuso, M.E.; Stephensen, D.; Shapiro, A.D.; Blanchette, V. Haemophilia. Nat. Rev. Dis. Prim. 2021, 7, 45.
- Castaman, G.; Matino, D. Hemophilia A and B: Molecular and clinical similarities and differences. Haematologica 2019, 104, 1702–1709.
- Perrin, G.Q.; Herzog, R.W.; Markusic, D.M. Update on clinical gene therapy for hemophilia. Blood 2019, 133, 407–414.
- Peyvandi, F.; Garagiola, I.; Young, G. The past and future of haemophilia: Diagnosis, treatments, and its complications. Lancet 2016, 388, 187–197.
- Pipe, S.W.; Gonen-Yaacovi, G.; Segurado, O.G. Hemophilia A gene therapy: Current and next-generation approaches. Expert Opin. Biol. Ther. 2022, 1–17.
- Everett, L.A.; Cleuren, A.; Khoriaty, R.N.; Ginsburg, D. Murine coagulation factor VIII is synthesized in endothelial cells. Blood 2014, 123, 3697–3705.
- Fahs, S.A.; Hille, M.T.; Shi, Q.; Weiler, H.; Montgomery, R.R. A conditional knockout mouse model reveals endothelial cells as the principal and possibly exclusive source of plasma factor VIII. Blood 2014, 123, 3706–3713.
- Shahani, T.; Covens, K.; Lavend’Homme, R.; Jazouli, N.; Sokal, E.; Peerlinck, K.; Jacquemin, M. Human liver sinusoidal endothelial cells but not hepatocytes contain factor VIII. J. Thromb. Haemost. 2014, 12, 36–42.
- Blanchette, V.S.; Key, N.S.; Ljung, L.R.; Manco-Johnson, M.J.; van den Berg, H.M.; Srivastava, A. Definitions in hemophilia: Communication from the SSC of the ISTH. J. Thromb. Haemost. 2014, 12, 1935–1939.
- Collins, P.W.; Blanchette, V.S.; Fischer, K.; Björkman, S.; Oh, M.; Fritsch, S.; Schroth, P.; Spotts, G.; Astermark, J.; Ewenstein, B.; et al. Break-through bleeding in relation to predicted factor VIII levels in patients receiving prophylactic treatment for severe hemophilia A. J. Thromb. Haemost. 2009, 7, 413–420.
- Uijl, I.E.M.D.; Fischer, K.; Van Der Bom, J.G.; Grobbee, D.E.; Rosendaal, F.R.; Plug, I. Analysis of low frequency bleeding data: The association of joint bleeds according to baseline FVIII activity levels. Haemophilia 2011, 17, 41–44.
- Lisowski, L.; Staber, J.M.; Wright, J.F.; Valentino, L.A. The intersection of vector biology, gene therapy, and hemophilia. Res. Pract. Thromb. Haemost. 2021, 5, e12586.
- Uijl, I.E.M.D.; Bunschoten, E.P.M.; Roosendaal, G.; Schutgens, R.E.G.; Biesma, D.H.; Grobbee, D.E.; Fischer, K. Clinical severity of haemophilia A: Does the classification of the 1950s still stand? Haemophilia 2011, 17, 849–853.
- Doshi, B.S.; Arruda, V.R. Gene therapy for hemophilia: What does the future hold? Ther. Adv. Hematol. 2018, 9, 273–293.
- Mancuso, M.E.; Mahlangu, J.N.; Pipe, S.W. The changing treatment landscape in haemophilia: From standard half-life clotting factor concentrates to gene editing. Lancet 2021, 397, 630–640.
- Mannucci, P.M.; Tuddenham, E.G. The Hemophilias—From Royal Genes to Gene Therapy. N. Engl. J. Med. 2001, 344, 1773–1779.
- Peyvandi, F.; Garagiola, I. Clinical advances in gene therapy updates on clinical trials of gene therapy in haemophilia. Haemophilia 2019, 25, 738–746.
- Nathwani, A.C.; Tuddenham, E.G.D. Haemophilia, the journey in search of a cure. 1960–2020. Br. J. Haematol. 2020, 191, 573–578.
- Olgasi, C.; Borsotti, C.; Merlin, S.; Bergmann, T.; Bittorf, P.; Adewoye, A.B.; Wragg, N.; Patterson, K.; Calabria, A.; Benedicenti, F.; et al. Efficient and safe correction of hemophilia A by lentiviral vector-transduced BOECs in an implantable device. Mol. Ther. Methods Clin. Dev. 2021, 23, 551–566.
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Investig. 2008, 118, 3132–3142.
- Hernández, R.J.; Calabria, A.; Sanvito, F.; De Mattia, F.; Farinelli, G.; Scala, S.; Visigalli, I.; Carriglio, N.; De Simone, M.; Vezzoli, M.; et al. Hematopoietic Tumors in a Mouse Model of X-linked Chronic Granulomatous Disease after Lentiviral Vector-Mediated Gene Therapy. Mol. Ther. 2021, 29, 86–102.
- Schlimgen, R.; Howard, J.; Wooley, D.; Thompson, M.; Baden, L.R.; Yang, O.O.; Christiani, D.C.; Mostoslavsky, G.; Diamond, D.V.; Duane, E.G.; et al. Risks Associated with Lentiviral Vector Exposures and Prevention Strategies. J. Occup. Environ. Med. 2016, 58, 1159–1166.
- Kumar, S.R.; Markusic, D.M.; Biswas, M.; High, K.A.; Herzog, R.W. Clinical development of gene therapy: Results and lessons from recent successes. Mol. Ther. Methods Clin. Dev. 2016, 3, 16034.
- Colella, P.; Ronzitti, G.; Mingozzi, F. Emerging Issues in AAV-Mediated In Vivo Gene Therapy. Mol. Ther. Methods Clin. Dev. 2018, 8, 87–104.
- Mendell, J.R.; Al-Zaidy, S.A.; Rodino-Klapac, L.R.; Goodspeed, K.; Gray, S.J.; Kay, C.N.; Boye, S.L.; Boye, S.E.; George, L.A.; Salabarria, S.; et al. Current Clinical Applications of In Vivo Gene Therapy with AAVs. Mol. Ther. 2021, 29, 464–488.
- Dunbar, C.E.; High, K.A.; Joung, J.K.; Kohn, D.B.; Ozawa, K.; Sadelain, M. Gene therapy comes of age. Science 2018, 359, eaan4672.
- Goverdhana, S.; Puntel, M.; Xiong, W.; Zirger, J.; Barcia, C.; Curtin, J.; Soffer, E.; Mondkar, S.; King, G.; Hu, J.; et al. Regulatable gene expression systems for gene therapy applications: Progress and future challenges. Mol. Ther. 2005, 12, 189–211.
- Li, C.; Samulski, R.J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 2020, 21, 255–272.
- Li, H.; Malani, N.; Hamilton, S.R.; Schlachterman, A.; Bussadori, G.; Edmonson, S.E.; Shah, R.; Arruda, V.R.; Mingozzi, F.; Wright, J.F.; et al. Assessing the potential for AAV vector genotoxicity in a murine model. Blood 2011, 117, 3311–3319.
- Nguyen, G.N.; Everett, J.K.; Kafle, S.; Roche, A.M.; Raymond, H.E.; Leiby, J.; Wood, C.; Assenmacher, C.-A.; Merricks, E.P.; Long, C.T.; et al. A long-term study of AAV gene therapy in dogs with hemophilia A identifies clonal expansions of transduced liver cells. Nat. Biotechnol. 2021, 39, 47–55.
- Rosas, L.E.; Grieves, J.L.; Zaraspe, K.; La Perle, K.; Fu, H.; McCarty, D.M. Patterns of scAAV Vector Insertion Associated with Oncogenic Events in a Mouse Model for Genotoxicity. Mol. Ther. 2012, 20, 2098–2110.
- Rangarajan, S.; Walsh, L.; Lester, W.; Perry, D.; Madan, B.; Laffan, M.; Yu, H.; Vettermann, C.; Pierce, G.F.; Wong, W.Y.; et al. AAV5–Factor VIII Gene Transfer in Severe Hemophilia A. N. Engl. J. Med. 2017, 377, 2519–2530.
- Pasi, K.J.; Laffan, M.; Rangarajan, S.; Robinson, T.M.; Mitchell, N.; Lester, W.; Symington, E.; Madan, B.; Yang, X.; Kim, B.; et al. Persistence of haemostatic response following gene therapy with valoctocogene roxaparvovec in severe haemophilia A. Haemophilia 2021, 27, 947–956.
- Pasi, K.J.; Rangarajan, S.; Mitchell, N.; Lester, W.; Symington, E.; Madan, B.; Laffan, M.; Russell, C.B.; Li, M.; Pierce, G.F.; et al. Multiyear Follow-up of AAV5-hFVIII-SQ Gene Therapy for Hemophilia A. N. Engl. J. Med. 2020, 382, 29–40.
- George, L.A.; Monahan, P.E.; Eyster, M.E.; Sullivan, S.K.; Ragni, M.V.; Croteau, S.E.; Rasko, J.E.; Recht, M.; Samelson-Jones, B.J.; MacDougall, A.; et al. Multiyear Factor VIII Expression after AAV Gene Transfer for Hemophilia A. N. Engl. J. Med. 2021, 385, 1961–1973.
- Sullivan, S.; Barrett, J.; Drelich, D.; Tarantino, M.; MacDougall, A.; Joseney-Antoine, M.; Wachtel, K.; Jaworski, K.; Curran, M.; Kuranda, K. SPK-8016: Preliminary results from a phase 1/2 clinical trial of gene therapy for hemophilia A. Haemophilia 2021, 136, 129–130.
- Visweshwar, N.; Harrington, T.J.; Leavitt, A.D.; Konkle, B.A.; Giermasz, A.; Stine, K.; Rupon, J.; Di Russo, G.; Tseng, L.-J.; Resa, M.D.L.A.; et al. Updated Results of the Alta Study, a Phase 1/2 Study of Giroctocogene Fitelparvovec (PF-07055480/SB-525) Gene Therapy in Adults with Severe Hemophilia a. Blood 2021, 138, 564.
- European Haemophilia Consortium; National Hemophilia Foundation; World Federation of Hemophilia. FDA Places the Pfizer/Sangamo Therapeutics Phase 3 AFFINE Haemophilia a Gene Therapy Study on Clinical Hold. Available online: https://www.hemophilia.org/sites/default/files/document/files/Pfizer-GT-pause-clean.pdf (accessed on 22 November 2021).
- Chapin, J.; Allen, G.; Alvarez-Roman, M.; Ayash-Rashkovsky, M.; Jaime, F.; Maggiore, C.; Mingot-Castellano, M.; Rajavel, K.; Rauch, A.; Susen, S. Results from a phase 1/2 safety and dose escalation study of TAK-754, an AAV8 vector with a codon-optimized B-domain-deleted factor VIII transgene in severe hemophilia A. Haemophilia 2021, 27, 122.
- Pipe, S.; Hay, C.; Sheehan, J.; Lissitchkov, T.; Leebeek, F.; Coppens, M.; Detering, E.; Ribeiro, S.; Vanevski, K. First-in-human gene therapy study of AAVhu37 capsid vector technology in severe hemophilia A: Safety and FVIII activity results. Res. Pract. Thromb. Haemost. 2020, 4, 27–28.
- Pipe, S.W.; Sheehan, J.P.; Coppens, M.; Eichler, H.; Linardi, C.; Wiegmann, S.; Hay, C.R.; Lissitchkov, T. First-in-Human Dose-Finding Study of AAVhu37 Vector-Based Gene Therapy: BAY 2599023 Has Stable and Sustained Expression of FVIII over 2 Years. Blood 2021, 138, 3971.
- Nathwani, A.C.; Tuddenham, E.; Chowdary, P.; McIntosh, J.; Lee, D.; Rosales, C.; Phillips, M.; Pie, J.; Junfang, Z.; Meagher, M.M. GO-8: Preliminary results of a Phase I/II dose escalation trial of gene therapy for haemophilia A using a novel human factor VIII variant. Blood 2018, 132, 489.
- Nathwani, A.C. Gene therapy for hemophilia. Hematol. Am. Soc. Hematol. Educ. Program 2019, 2019, 1–8.
- Zhang, F.; Yan, X.; Li, M.M.; Hua, B.; Xiao, X.; Monahan, P.E.; Sun, J. Exploring the Potential Feasibility of Intra-Articular Adeno-Associated Virus-Mediated Gene Therapy for Hemophilia Arthropathy. Hum. Gene Ther. 2020, 31, 448–458.
- Nathwani, A.C.; Tuddenham, E.G.D.; Rangarajan, S.; Rosales, C.; McIntosh, J.; Linch, D.C.; Chowdary, P.; Riddell, A.; Pie, A.J.; Harrington, C.; et al. Adenovirus-Associated Virus Vector–Mediated Gene Transfer in Hemophilia B. N. Engl. J. Med. 2011, 365, 2357–2365.
- Mingozzi, F.; High, K.A. Immune responses to AAV vectors: Overcoming barriers to successful gene therapy. Blood 2013, 122, 23–36.
- Lenting, P.J.; van Mourik, J.A.; Mertens, K. The life cycle of coagulation factor VIII in view of its structure and function. Blood 1998, 92, 3983–3996.
- Lisowski, L.; Dane, A.P.; Chu, K.; Zhang, Y.; Cunningham, S.C.; Wilson, E.M.; Nygaard, S.; Grompe, M.; Alexander, I.E.; Kay, M.A. Selection and evaluation of clinically relevant AAV variants in a xenograft liver model. Nature 2014, 506, 382–386.
- Nathwani, A.C.; Gray, J.T.; Ng, C.Y.C.; Zhou, J.; Spence, Y.; Waddington, S.; Tuddenham, E.G.D.; Kemball-Cook, G.; McIntosh, J.; Boon-Spijker, M.; et al. Self-complementary adeno-associated virus vectors containing a novel liver-specific human factor IX expression cassette enable highly efficient transduction of murine and nonhuman primate liver. Blood 2006, 107, 2653–2661.
- Mahlangu, J.; Oldenburg, J.; Paz-Priel, I.; Negrier, C.; Niggli, M.; Mancuso, M.E.; Schmitt, C.; Jiménez-Yuste, V.; Kempton, C.; Dhalluin, C.; et al. Emicizumab Prophylaxis in Patients Who Have Hemophilia A without Inhibitors. N. Engl. J. Med. 2018, 379, 811–822.
- Ozelo, M.C.; Mahlangu, J.; Pasi, K.J.; Giermasz, A.; Leavitt, A.D.; Laffan, M.; Symington, E.; Quon, D.V.; Wang, J.-D.; Peerlinck, K.; et al. Valoctocogene Roxaparvovec Gene Therapy for Hemophilia A. N. Engl. J. Med. 2022, 386, 1013–1025.
- Verdera, H.C.; Kuranda, K.; Mingozzi, F. AAV Vector Immunogenicity in Humans: A Long Journey to Successful Gene Transfer. Mol. Ther. 2020, 28, 723–746.
- Weber, T. Anti-AAV Antibodies in AAV Gene Therapy: Current Challenges and Possible Solutions. Front. Immunol. 2021, 12, 658399.
- Stanford, S.; Pink, R.; Creagh, D.; Clark, A.; Lowe, G.; Curry, N.; Pasi, J.; Perry, D.; Fong, S.; Hayes, G.; et al. Adenovirus-associated antibodies in UK cohort of hemophilia patients: A seroprevalence study of the presence of adenovirus-associated virus vector-serotypes AAV5 and AAV8 neutralizing activity and antibodies in patients with hemophilia A. Res. Pract. Thromb. Haemost. 2019, 3, 261–267.
- Batty, P.; Lillicrap, D. Hemophilia Gene Therapy: Approaching the First Licensed Product. HemaSphere 2021, 5, e540.
- Leavitt, A.D.; Konkle, B.A.; Stine, K.; Visweshwar, N.; Harrington, T.J.; Giermasz, A.; Arkin, S.; Fang, A.; Plonski, F.; Smith, L. Updated follow-up of the Alta Study, a phase 1/2 study of giroctocogene fitelparvovec (SB-525) gene therapy in adults with severe hemophilia A. Blood 2020, 136, 12.
- Sabatino, D.E.; Lange, A.M.; Altynova, E.S.; Sarkar, R.; Zhou, S.; Merricks, E.P.; Franck, H.G.; Nichols, T.C.; Arruda, V.R.; Kazazian, H.H., Jr. Efficacy and Safety of Long-term Prophylaxis in Severe Hemophilia A Dogs Following Liver Gene Therapy Using AAV Vectors. Mol. Ther. 2011, 19, 442–449.
- Leebeek, F.W.G.; Miesbach, W. Gene therapy for hemophilia: A review on clinical benefit, limitations, and remaining issues. Blood 2021, 138, 923–931.
- Konkle, B.A.; Recht, M.; Hilger, A.; Marks, P. The critical need for postmarketing surveillance in gene therapy for haemophilia. Haemophilia 2021, 27, 126–131.
- Pierce, G.F.; Kaczmarek, R.; Noone, D.; O’Mahony, B.; Page, D.; Skinner, M.W. Gene therapy to cure haemophilia: Is robust scientific inquiry the missing factor? Haemophilia 2020, 26, 931–933.
- Miesbach, W.; Klamroth, R. The Patient Experience of Gene Therapy for Hemophilia: Qualitative Interviews with Trial Patients. Patient Prefer. Adherence 2020, 14, 767–770.
- Woollard, L.; Gorman, R.; Rosenfelt, D.J. Improving patient informed consent for haemophilia gene therapy: The case for change. Ther. Adv. Rare Dis. 2021, 2, 26330040211047244.
- Sidonio, R.F.; Pipe, S.W.; Callaghan, M.U.; Valentino, L.A.; Monahan, P.E.; Croteau, S.E. Discussing investigational AAV gene therapy with hemophilia patients: A guide. Blood Rev. 2021, 47, 100759.


