The standard of care for hemophilia A has been intravenous administration of exogenous factor VIII (FVIII), either as prophylaxis or episodically. Gene therapy is a treatment strategy used to repair or provide a functional copy of a gene that is either absent or expressed as a non-functional protein [4,28,29,30]. Treating a genetic disease with gene therapy requires that the transgene (or its protein product) be delivered to the physiologically relevant target tissue or tissues, be stably expressed, and not interfere with the functional integrity of the cells in these tissues [31,32,33]. The ultimate goal of gene therapy for patients with hemophilia A is the production of a treatment that is given as a one-time infusion and that allows adequate long-term expression of the deficient FVIII, with the maintenance of steady-state plasma FVIII concentrations. This would minimize (or ideally eliminate) bleeding episodes and thereby decrease the patient and societal burden of the disease [34,35,36,37].
1. Introduction
Hemophilia is a bleeding disorder that includes two distinct genetic disorders caused by missing or defective clotting factor VIII (FVIII; hemophilia A) or clotting factor IX (hemophilia B) [
1,
2,
3,
4,
5,
6].
FVIII is a large protein that is endogenously produced, primarily in the liver by sinusoidal endothelial cells (rather than hepatocytes) [
4,
7,
8,
9]. Disease severity in hemophilia A is classified according to the plasma level of FVIII activity [
2,
10]. The severe form of hemophilia A is defined as a FVIII activity level <1% of normal, the moderate form as a FVIII level of 1–5% of normal, and the mild form as a FVIII level >5% to <40% of normal [
10]. Endogenous FVIII activity levels of <1 IU/dL (1%) present an increased risk of spontaneous and trauma-related bleeding events, including bleeding into the joints that leads to hemophilic arthropathy. In contrast, an endogenous FVIII activity level >1 IU/dL is associated with a low risk of breakthrough bleeding and progressive joint destruction [
11,
12].
Currently available therapeutic options for hemophilia A (primarily, intravenous administration of exogenous FVIII) are associated with several limitations and there is an unmet need for improved care in these patients. Gene therapy represents an emerging effective long-term treatment modality for hemophilia that potentially bypasses the complications of other therapies, reducing the number of breakthrough bleeding events and the need for frequent infusions, thereby improving patient health-related quality of life (QoL) [
4]. Despite its potential, several issues remain to be fully clarified for hemophilia A gene therapy, including patient selection criteria, the durability and variability of transgene expression, and long-term safety. In addition, the complexity and potential complications of prescribing, administering, and monitoring patients undergoing gene therapy necessitate specific considerations for the centers that will offer this therapy, i.e., expert ‘hubs’, in which a multidisciplinary team will be required to ensure maximum benefit from therapy and optimal levels of treatment and care.
2. Gene Therapy in Hemophilia A
After more than three decades of investigation, gene therapy in hemophilia A has moved beyond proof of concept, toward a realistic expectation that the first product may soon be available. Indeed, such a gene therapy is likely to be approved in Europe in 2022.
Hemophilia A is an ideal candidate for gene therapy [
32,
38,
39,
40,
41,
42]. Its well-characterized monogenic nature of inheritance allows for the correction of a single gene to provide symptomatic relief [
32,
43]. In hemophilia A, gene transfer strategies employing adeno-associated viral (AAV) vectors to target liver hepatocytes are the closest to regulatory approval. Lentiviral vectors (LVs) targeting hematopoietic stem and progenitor cells ex vivo have shown promise in animal models [
44] and have advanced into clinical testing [
6]; however, lentiviral vectors integrate into DNA and may potentially induce oncogenesis [
45,
46,
47].
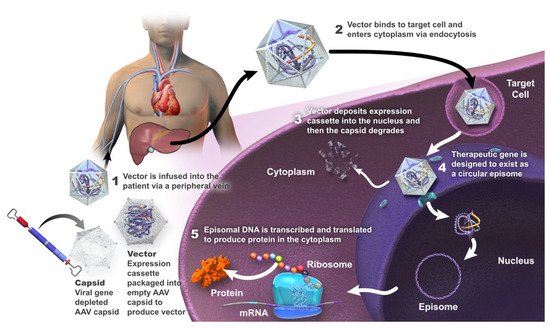
To date, AAV vectors have demonstrated the greatest clinical success for in vivo gene delivery [
29,
48,
49] (
Figure 1).
Figure 1. Summary of mechanism of action of gene therapy using an adeno-associated viral (AAV) vector.
AAV vectors are engineered from a non-pathogenic, non-enveloped linear, single-stranded DNA parvovirus; wild-type AAV is naturally replication-defective and requires a helper virus for replication [
4,
50,
51,
52]. These vectors are minimally integrative and associated with a low risk of insertional mutagenesis [
48,
53,
54,
55]. The host transcription machinery transcribes the transgene into mRNA, which is then translated into the protein of interest [
6]. It is considered that the genome of the recombinant AAV vector does not undergo site-specific integration in the host DNA or alter the genetic process, but is largely episomal in the nucleus of transduced cells [
48].
2.1. Valoctocogene Roxaparvovec
Valoctocogene roxaparvovec is a recombinant codon-optimized AAV5 vector that expresses the SQ variant of B-domain-deleted human FVIII with a hybrid liver-specific transcription promoter. It is likely to be the first gene therapy to be approved as, in June 2022, EMA recommended granting conditional marketing authorization in the EU. The expression cassette is inserted between two AAV serotype 2 inverted terminal repeats [
69]. The non-enveloped icosahedral AAV5 capsid delivers the transgene predominantly to the liver [
69]. The vector itself is manufactured using a baculovirus–
Spodoptera frugiperda (Sf9) insect–cell production system [
69].
In an ongoing phase ½ study (NCT02576795), an infusion of valoctocogene roxaparvovec was administered to 15 adults with severe hemophilia A (FVIII ≤ 1 IU/dL) at doses of 6 × 10
12 viral genome (vg)/kg (n = 1), 2 × 10
13 vg/kg (n = 1), 6 × 10
13 vg/kg (n = 7) or 4 × 10
13 vg/kg (n = 6) [
64,
65,
69]. Four of seven and three of six participants in the 6 × 10
13 vg/kg and 4 × 10
13 vg/kg cohorts, respectively, maintained median FVIII levels > 5 IU/dL at 4 and 5 years after administration [
64]. Individuals in the 6 × 10
13 vg/kg cohort had a mean ABR of 0.7 treated bleeds/year during year 5, resulting in a cumulative mean ABR of 0.8 treated bleeds/year from week 5 onward (95% reduction from baseline) [
64]. Individuals in the 4 × 10
13 vg/kg cohort had a mean ABR of 1.7 treated bleeds/year during year 4, resulting in a cumulative mean ABR of 1.0 treated bleeds/year from week 5 onward, a 92% reduction from baseline [
64]. QoL was maintained in patients receiving the higher dose [
64]. The most common adverse events were transient, asymptomatic, and mild-to-moderate ALT elevations [
64].
2.2. Dirloctocogene Samoparvovec
Dirloctocogene samoparvovec (SPK-8011) is a recombinant-AAV vector consisting of SPK200 (a bioengineered capsid derived from AAV3 (specifically, subtype LK03)) with a liver-specific, truncated transthyretin enhancer and promoter, a synthetic intron sequence, and codon-optimized FVIII cDNA encoding FVIII-SQ and manufactured with transient triple transfection of human embryonic kidney cells (HEK293 cells) [
58].
In a phase ½ trial (NCT03003533/NCT03432520), 18 men with hemophilia A were enrolled in four dose cohorts; the lowest-dose cohort received 5 × 10
11 vg/kg and the highest-dose cohort received 2 × 10
12 vg/kg, with glucocorticoids administered in cases of suspected immune response [
58]. Two participants lost all FVIII expression because of an anti-AAV capsid cellular immune response that was not sensitive to immune suppression. In the remaining 16 men, FVIII expression was maintained, with 12 men followed for >2 years. These men had a 91.5% reduction in ABR after dirloctocogene samoparvovec administration. Elevated ALT levels were seen in seven study participants, the majority of which were mild, with the exception of a grade 2 elevation in one participant that required hospitalization for treatment. Adverse events related to glucocorticoids were reported in four participants. No cases of FVIII inhibitory antibody development were reported.
2.3. SPK-8016
A phase ½, open-label, non-randomized, dose-finding study is investigating the efficacy of SPK-8016 in adult men with clinically severe hemophilia A who have not developed FVIII inhibitors and had no AAV neutralizing antibodies (NCT03734588) [
70]. In three patients, there was evidence for unstable FVIII:C and cellular immunity against AAV. Patients were put on daily oral corticosteroids at weeks 3–7 for 43–38 weeks. Preliminary results demonstrated sustained FVIII levels (6.2–21.8%) after 52 weeks in four men who received SPK-8016 at a dose of 5 × 10
11 vg/kg. An optimization of the immunomodulatory regimen is being developed with the potential for obtaining clinically meaningful FVIII activity using low vector doses [
70].
2.4. Giroctocogene Fitelparvovec
Giroctocogene fitelparvovec comprises an AAV serotype 6 vector (AAV2/6) encoding the cDNA for B domain-deleted human FVIII. This gene therapy has been investigated in a phase ½ trial (NCT03061201) in which 11 patients were each given four ascending doses: 9 × 10
11, 2 × 10
12, 1 × 10
13, and 3 × 10
13 vg/kg (n = 2, 2, 2 and 5 patients, respectively) [
71]. Four patients in the 3 × 10
13 vg/kg cohort have available data, which demonstrated mean FVIII activity maintained in the mild to normal range (30.9% measured by CCA; 46.4% measured by one-stage assay) to week 104. The ABR was zero for the first-year post-infusion and 0.9 throughout the follow-up, and two patients experienced a total of three bleeding events (two traumatic, one unknown; one in a target joint) that necessitated treatment with exogenous FVIII. No patients have resumed prophylaxis. Common treatment-related adverse events were elevated liver enzyme levels (ALT n = 5/11 patients, and AST n = 3/11), pyrexia (n = 3/11), and tachycardia (n = 2/11). Four patients in the 3 × 10
13 vg/kg cohort had an ALT elevation that required >7 days of corticosteroid treatment, but these were managed with a tapering course of corticosteroids, and FVIII activity was maintained. No patients developed inhibitors to FVIII.
Giroctocogene fitelparvovec was being evaluated in a phase 3 study in men with moderately severe to severe hemophilia A (AFFINE; NCT04370054) [
57], but the US Food and Drug Administration has placed this study on hold until proposed protocol amendments have been implemented, due to FVIII levels being >150% in some participants [
57].
2.5. TAK-754
A phase ½ trial (NCT03370172) is currently evaluating TAK-754, a modified AAV serotype 8 gene therapy, in four men with severe hemophilia A [
56]. Participants were treated in two ascending dose cohorts (2 × 10
12 capsid particles [cp]/kg and 6 × 10
12 cp/kg) [
56]. FVIII expression declined significantly during the tapering of corticosteroids, and three of the four patients have resumed FVIII replacement therapy after ≥10 months’ follow-up. The utilization of corticosteroids did not prevent the loss of transgene expression. The reason behind transcript expression loss is being investigated [
56].
2.6. BAY 2599023
BAY 2599023 (AAVhu37.hFVIIIco) comprises an AAV vector with a capsid serotype hu37 (AAVhu37), and a genome that directs expression of a codon-optimized B-domain-deleted human FVIII under the control of a liver-specific promoter/enhancer combination [
67].
A phase ½ study (NCT03588299) is being conducted in adult males who have severe hemophilia A with no history of FVIII inhibitors, no detectable immunity to the AAVhu37 capsid, and ≥150 exposure days to FVIII products [
67,
68]. Nine patients have received a single infusion of BAY 2599023 at doses of 0.5, 1.0, and 2.0 × 10
13 gene copies/kg (n = 2, 2, and 5, respectively) [
67,
68].
2.7. AAV8-HLP-hFVIII-V3
AAV-HLP-hFVIII-V3 encodes a 17-amino-acid peptide with six N-linked glycosylation motifs from the human FVIII B domain, pseudotyped with AAV serotype 8 capsid, and has been shown to mediate three-fold higher FVIII expression compared with valoctocogene roxaparvovec in an animal model [
61]. In a phase ½ study (NCT03001830), three men with severe hemophilia A received a single infusion of AAV8-HLP-hFVIII-V3 [
61], with one man receiving a dose of 6 × 10
11 vg/kg and the subsequent two patients each receiving a dose of 2 × 10
12 vg/kg. The patients were followed up for 13–47 weeks after vector administration. FVIII activity levels were >5% in all three men, with normalization of FVIII levels in one patient. Serum ALT levels were elevated in two of the patients between 4 and 6 weeks after gene transfer but this responded to corticosteroids, with no loss of transgene expression. No study participants have developed FVIII inhibitors.
3. Opportunities and Challenges of Gene Therapy in Hemophilia A
3.1. Opportunities
Gene therapy is a disease-transforming therapy that has the potential to become the new standard for the treatment of patients with hemophilia A [72]. It may enable the patient to achieve a sustained physiological level of endogenously produced FVIII protein that could provide effective prophylaxis without the need for exogenous factor replacement therapy [40,42]. Such continuous endogenous expression of physiological levels of FVIII can be expected to eliminate breakthrough bleeding and micro-hemorrhages [72]. Gene therapy may, thus, provide protection against further bleeding-induced joint damage [73]. Given that even minimal increases in FVIII levels can lead to significantly improved bleeding outcomes, it is expected that gene therapy will provide a resultant improvement in the QoL of the individual with hemophilia A [38,72].
3.2. Challenges
Successful gene therapy was first reported, in 2011, with intravenously administered AAV-based liver-directed gene therapy in patients with severe hemophilia B [74]. In contrast, gene therapy directed at hemophilia A has lagged behind that directed at hemophilia B, largely due to the limitations inherent in the size of the FVIII gene and the structure of FVIII, problems achieving therapeutic levels of the transgenic protein, and cellular immune responses to the capsid of the AAV vector [75] (also identified in trials of therapy for hemophilia B, although to a lesser extent), as well as issues related to the duration and variability of this form of therapy [1,4,6].
FVIII Structure
The gene for FVIII is located at the tip of the long arm of the X chromosome. At over 180 kb, it is one of the largest known genes. It comprises 26 exons, which encode a polypeptide chain of 2351 amino acids (19 signal peptides and a 2332-amino-acid mature polypeptide chain). Factor VIII is composed of six domains, A1-A2-B-A3-C1-C2, with three acidic regions (α1-3) bordering the A domains. The A domains are each approximately 330 residues in length, the B domain approximately 900 residues, and the C domains each approximately 150 residues [
76].
FVIII Expression
The target cell for FVIII gene therapy is the hepatocyte, which is not the native cell for the endogenous FVIII synthesis [
7,
8]. Nevertheless, expression of FVIII in hepatocytes generates a functional and physiological protein that has restored hemostasis in animal models and clinical studies [
81,
82]. In contrast, other currently available therapies involve the administration of exogenous FVIII by infusion or the subcutaneous administration of an FVIII-mimicking compound [
2,
14].
Infusion-Related Adverse Reactions
Infusion-related adverse reactions can occur with gene therapy for hemophilia A, but are generally infrequent, resolve with treatment and/or slowing or pausing the infusion, and do not usually prevent completion of the infusion. In a phase ½ study (NCT02576795) of valoctocogene roxaparvovec (an AAV5-based gene therapy encoding human B domain-deleted FVIII), one patient was hospitalized due to grade 2 pyrexia with myalgia and headache that occurred within 24 h after the infusion; the symptoms resolved within 48 h after treatment with acetaminophen (paracetamol) [
65]. In a phase 3 study of valoctocogene roxaparvovec (NCT03370913), seven study participants (5.2%) had systemic hypersensitivity and three reported serious infusion-related reactions (maculopapular rash and presyncope; anaphylactic reaction; and hypersensitivity reaction) during or shortly after the infusion [
63]. However, the infusion reactions were managed by slowing or pausing the infusion and administering supportive medications (e.g., antihistamines, antipyretics, or glucocorticoids), such that all participants completed the infusion.
Anti-AAV Neutralizing Antibodies
Exposure to wild-type AAV results in priming of the body’s immune system against the virus, with the development of both humoral and T-cell immunity that can neutralize the vector and reduce treatment efficacy [
85,
86]. At present, patient selection in gene therapy trials in hemophilia A includes screening for pre-existing immunity to identify and enroll patients who are negative for anti-AAV capsid neutralizing antibodies [
6,
87].
Elevation of Liver Enzymes
AAV-based gene therapy may induce a degree of hepatotoxicity, which is usually transient and asymptomatic. Increased levels of serum ALT are commonly seen. The mechanism of this toxicity has not been clearly established but could be related to the development of an anti-AAV capsid peptide cytotoxic T cell response, endoplasmic reticulum stress, and associated hepatocyte apoptosis due to high expression of FVIII, or a direct effect of vector particle load [
80]. Clinical studies in patients with hemophilia A have reported increases in ALT level in a high proportion of patients [
56,
58,
60,
61,
63,
65,
67,
69,
70]. In a phase ½ study of valoctocogene roxaparvovec in patients with hemophilia A, an increase in ALT was reported in 11 of the 15 study participants (73.3%; 14 events). The increases were not generally associated with loss of FVIII activity or a T-cell immune response to viral capsid peptides [
65]. All elevations in ALT were mild (13 events grade 1 and 1 event grade 2), non-serious and transient, and there were no symptoms or sequelae suggestive of clinically significant hepatocyte injury or liver dysfunction [
65]. There were no consistent cellular immune responses to FVIII or AAV5 capsid detected in any participant [
65].
Oncogenesis
The risk of genomic insertional mutagenesis after AAV-mediated gene transfer is considered to be low due to the episomic nature of the transinfected cDNA [
6]. Durable FVIII expression has been observed in an animal model of severe hemophilia A, with no late-toxicity or oncogenesis [
94]. A study in hemophilia dogs treated with AAV8 or AAV9 vectors expressing canine FVIII and followed for 10 years did not find any evidence of hepatic tumors or altered liver function, although there were some AAV integration events in genomic DNA and clonal expansion of cells that harbored vector integration in genes potentially associated with growth control [
54].
Durability and Variability of Transgene Expression
The clinical trials completed so far have not answered fully all issues related to gene therapy in hemophilia A. Open questions remain as to why expression levels of FVIII decrease over time and why there is intra- and inter-patient variability [
83]. It is yet to be determined whether the vector serotype, manufacturing process, expression of FVIII in hepatocytes, FVIII protein unfolding, episomal structural changes, or other factors contribute to this decline/variability in FVIII levels [
1]. The impact of liver growth and hepatocyte turnover on AAV vector genome persistence, and the efficacy and durability of factor expression in adults, adolescents, and pediatric groups is also still unclear [
97]. Variability may or may not be related to durability [
98]. Identifying the causes of inter- and intra-individual variability needs to be a research priority, as finding the root causes of the variability might permit potential mitigants to be identified [
98].
Patient Expectations
Gene therapy has the potential to change the quality of life of patients with hemophilia A by reducing the frequency of bleeding events, thereby allowing patients to lead lives that are less impacted by the disease [
83,
101,
102]. However, it is important that patient expectations surrounding their eligibility, access to treatment, and treatment outcomes are managed appropriately, as this therapy may not be suitable for all patients [
83,
102,
103]. This is because there is variability in FVIII expression between patients, FVIII expression may not be durable in all patients, and some patients may be positive for anti-AAV capsid neutralizing antibodies and, therefore, not suitable candidates for gene therapy. People living with hemophilia A need to be fully informed regarding the potential benefits and risks of gene therapy [
102]. It will be essential that physicians pay close attention to what information they share with their patients, choose the optimum way to do so, and determine how to ensure sufficient patient understanding of the information provided [
102].
4. Conclusions
The current treatment of hemophilia A involves frequent intravenous injections and the risk of FVIII inhibitor development, which can negatively impact patient QoL. Gene therapy for patients with hemophilia A represents the potential for a single treatment that could allow for long-term expression of the deficient FVIII, with the maintenance of steady-state plasma FVIII concentrations, thereby minimizing bleeding episodes for the whole lifetime of the recipient and decreasing the burden of their disease. FVIII expressed through gene therapy is a physiological protein rather than a mimicking factor or an infused exogenous FVIII concentrate, and steady-state physiological plasma FVIII levels may avoid the deterioration of joint status.
Gene therapy for the management of hemophilia A has now moved beyond proof of concept, with the realistic expectation that it may become available to patients in European countries, including Italy, in 2022 [
49,
80]. It is likely that the first licensed product will be valoctocogene roxaparvovec, for which the longest duration efficacy and safety data (5 years) for any gene therapy currently under investigation are available. These data indicate sustained secretion of FVIII in patients who have received valoctocogene roxaparvovec, such that ABR and FVIII replacement therapy were minimized [
64].
Gene therapy could represent a paradigm shift, becoming a new reference standard in the treatment of patients with hemophilia A, enabling long-lasting treatment and improved clinical and patient-centered outcomes, including enhanced QoL, for many patients with this disease.
This entry is adapted from the peer-reviewed paper 10.3390/ijms231810228