 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Aleksander J. Nowak | + 1936 word(s) | 1936 | 2020-12-17 07:52:54 | | | |
| 2 | Catherine Yang | Meta information modification | 1936 | 2020-12-18 04:48:20 | | |
Video Upload Options
Molecular, pathological mechanisms of ALD principally root in the innate immunity system and are especially associated with enhanced functionality of the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway. NF-κB is an interesting and convoluted DNA transcription regulator, promoting both anti-inflammatory and pro-inflammatory gene expression. Thus, the abundancy of studies in recent years underlines the importance of NF-κB in inflammatory responses and the mechanistic stimulation of inner molecular motifs within the factor components.
1. Introduction
Ethanol (ethylic alcohol, EtOH) in the form of various and diversified beverages is one of the world’s most commonly consumed active ingredients in drinks, alongside caffeine [1]. Besides recreational and culturally related purposes, ethanol is also a ubiquitous chemical compound applied frequently as a disinfectant, an antiseptic, a solvent and an antidotal drug, as well as being used as a fuel base in the industry [2]. In the short term, alcohol exhibits multiple biological effects on the human organism, including a severe psychoactive stimulation (by disrupting signaling pathways in the brain, leading to euphoria, disinhibition or moods swings) [3], arrhythmia, ataxia or analgesia [4]. However, chronic EtOH consumption can lead to several complex diseases like alcoholic gastritis, alcoholic myopathy, alcoholic cardiomyopathy, alcoholic neuropathy, alcoholic pancreatitis and alcoholic liver disease (ALD) [5][6]. The latter one is a complex and progressive condition that comprises many stages regarding the advancement of the disease, where the treatment possibilities range from a simple alcoholic abstain in its milder forms to a liver transplantation in the acute and terminal phases [7]. The multi-layered pathogenesis of ALD is still not comprehensively understood, especially the direct alterations of signaling pathways in the liver cells, which stimulate an immune response and abundant release of pro-inflammatory factors, including tumor necrosis alpha (TNFα) and interleukin 1-beta (IL-1β) [8]. This leads to the change in expression levels of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), a crucial transcription factor complex [9]. NF-κB is an important signaling cascade regulator that controls the genes responsible for innate and adaptive immunological responses [10]. It is believed that chronic alcoholic disease induces heavy shifts in metabolism and signaling of both parenchymal and non-parenchymal liver cells, thus leading to accumulation of fatty acids in the liver and causing the disease progression [11]. This review focuses predominantly on a correlation between alcoholic liver disease and any alternations in function of the NF-κB regulatory complex during induced liver alcoholic injury in animal models. It is assumed that inner mechanisms of the ALD pathogenesis lie profoundly within the various liver cells via NF-κB signaling malfunctions [12].
2. NF-κB-Regulating Molecular Factors
The group of deubiquitinating enzymes (DUBs) is a paramount and vast class of protein superfamilies that altogether act as varied biochemical controllers by catalytically cleaving ubiquitin (Ub), a ubiquitous regulatory protein, from its molecular substrate targets [13]. The DUBs group consists of cysteine proteases and metalloproteases [14] that mechanistically process ubiquitin precursors to ubiquitin and detach the mono-ubiquitin or poly-ubiquitin chains from the corresponding proteins. All of these biochemical reactions are classified in the repertoire of post-translation modifications (PTMs) that frequently occur following protein biosynthesis in ribosomes [15]. A particularly fascinating DUB in regard to the NF-κB- and ALD-related discussion is a protein known as A20, discovered by Dixit et al. in the early 1990s [16]. A20, also known as tumor necrosis factor alpha-induced protein 3 (TNFAIP3 [17]), is expressed upon oxidative stress or inflammatory stimulation with, e.g., LPS, by a dozen of differentiated cell types, e.g., pancreatic cells [18], hepatocytes [19] or endothelial cells [16]. There is evidence that its influence on NF-κB is predominately negative, as it does contribute to the negative regulatory feedback loop that aims to refurbish the inflammatory condition to its unedited state, thus A20 functions as the NF-κB activity inhibitor [17]. Both of the A20 ubiquitin activities (E3 ubiquitination and DUB activity—the deubiquitinating activity from the amino-terminal ovarian tumor (OTU) domain) seem to have an enormous influence on NF-κB inhibition, although the exact explanation to this phenomenon still requires further investigation [20]. It was previously described that A20 removes lysine63-linked ubiquitin chains from an important TNF pathway receptor-interacting Ser-Thr protein kinase 1 (RIPK1) or NEMO from the IKK units and is capable of attaching lysine48-linked ubiquitin chains on RIPK1 or TNF-receptor, thus inducing the proteasome degradation process of marked targets [21]. However, there is experimental evidence published suggesting that the deubiquitinating activity of A20 might be marginal for NF-κB downregulation, as it presumably does not negatively affect inflammation progression in mice with inactivated DUB and E3 domains [22]. However, A20-deficient mice exhibit severe systemic inflammation and a high perinatal mortality rate [23]. To target the issue, Martens et al. generated recombined mice mutants, deficient, simultaneously, in the zinc finger 7 (ZnF7, poly-ubiquitin binding activity) and zinc finger 4 (ZnF4, the aforementioned E3 ligase activity) domains of A20 [24]. The study concluded in the acquirement of mice mutants exhibiting the same lethality levels and acute inflammation symptoms as the full-length A20-deficient mice [24]. Interestingly, the experimental group of Razani et al. achieved similar conclusions and results [25]. This underlies the pivotal role of A20 zinc finger domains and the wholesome ubiquitin-binding properties in inflammation protection. However, much has yet to be investigated, as it is unclear how A20 drives NF-κB regulation and inflammasome activation, especially in the setting of alcohol-induced liver injury. In terms of focused liver cell-based studies with a full-length A20 gene knockout (KO) approach, it was proven that A20 deficiency in HSCs causes lymphopenia, increased postnatal lethality and anemia in mice, which is a consequence of the loss of HSCs quiescence [26]. The A20-KO in hepatocytes results in chronic liver inflammation, as well as increased inflammation induction by pro-inflammatory stimulants like LPS or TNF, as well as enhanced susceptibility to HCC [19]. The protective properties of A20 towards HCC progression have been shown in another study, where A20 acts as an E3 Ub-ligase for a liver-type phosphofructokinase (PFKL) [27]. This leads to the inhibition of glycolysis and ceases cancerous proliferation, hence the evidence for the potential antitumor role of A20 emerges [27]. A20 was also suspected earlier to suppress HCC by its Twist1 inhibition activity [28]. To our knowledge, there are no investigations solely oriented to the role of A20 in the pathogenesis of ALD yet. Moreover, there is another important player in NF-κB inducible signaling also belonging to the DUBs, the so-called ubiquitin aldehyde binding 1 protein (OTUB-1, a member of the otubains family, the group of cysteine proteases [29]). It was reported already that OTUB-1 is an E2 ligase ubiquitin conjugated enzyme 13 (UBC13) [30] regulator, where UBC13 acts as a regulatory factor in the ring finger protein 168 (RFP168) signaling pathway [31], which participates in double-strand DNA breaks repair [32]. In consequence, OTUB-1 exhibits inhibitory properties on UBC13 [33]. Recently, it was also observed that OTUB-1 promotes NF-κB activation in dendritic cells during stimulated inflammatory conditions by lysine48 deubiquitination of UBC13 [34]. Additionally, there is evidence that OTUB-1 deficiency leads to a gradual degradation of the NF-κB precursor protein p100 to p52, which culminates in NF-κB activity induction [35]. These studies underline the OTUB-1 function as an important DUB-based NF-κB regulator, although a comprehensive experimental study in that regard should still be conducted.
A20 is the best-studied NF-κB-modulating DUB so far, however, it is important to mention another DUB regulator of NF-κB, the cylindromatosis tumor suppressor protein, CYLD [36]. The catalytic domain of CYLD, the ubiquitin specific protease domain (USP), is structurally also a cysteine protease, responsible for its DUB activity. CYLD, alongside A20, is suspected to remove lysine63-linked polyubiquitin chains from many targets involved in the transduction of the NF-κB signal, primarily NEMO and RIPK1 proteins [37]. CYLD exhibits affinity to different targets by binding adaptor proteins, e.g., p62, which guides it to the ubiquitinated ligands of interest [38]. Harhaj and Dixit described CYLD in a more investigative manner in the following articles [39][40]. OTULIN is a DUB that structurally solely constitutes the OTU domain with an N-terminal PUB-interacting motif, which is selective for binding methionine1-linked Ub chains in NF-κB transmitters [37][41]. Its systematic deficiency in expression causes OTULIN-related autoinflammatory syndrome (ORAS), which results in steatosis-alike liver inflammation [42]. However, not much is known about OTULIN and ORAS, as this protein functionality is still poorly understood and requires further investigations. Graphical interpretation of the DUBs functionality in modulating the NF-κB pathway and known interactions has been displayed in the Figure 2.
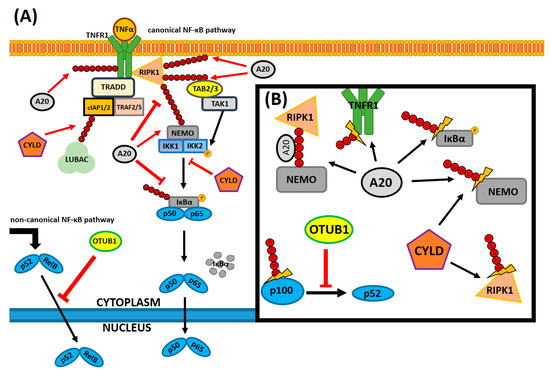
Figure 2. A simplified canonical NF-κB signaling pathway with its many deubiquitinating regulators and the molecular targets of those enzymes. Only a handful of all possible interactions between deubiquitinating enzymes (DUBs) and NF-κB pathway components are currently known and an even lesser amount of all the possible interactions are displayed here. (A) Canonical signaling in the NF-κB pathway consists of various receptors, where the signal is transduced through a cross-linked network of protein complexes and reaches the NF-κB heterodimeric subunits sequestrated in the cytosol. The signal from TNFα assembles the RIPK1 complex at the site of its binding with the receptor, which directs TAK1 to phosphorylate the IKK complex. In consequence, the IKK complex phosphorylates the inhibitor unit of sequestrated NF-κB proteins, which marks it for proteasomal degradation. After the proteolysis, the activated NF-κB heterodimer translocates to the nucleus and induces the transcription of target genes. Ubiquitin poly-chains, anchored by several amino acid residues (M1, K48 and K63, respectively, which, for the sake of the reading coherency, were not distinguished in this figure) to target proteins, fill a multitude of different roles: they create linkages and bridges between components and mark them for degradation or create additional signals. DUBs: A20 and CYLD are the key deubiquitinating regulators that not only remove ubiquitin chains from targets, but also bind to them. In addition, OTUB1’s novel role as an NF-κB inhibitor in the non-canonical pathway (not shown here). (B) Several Ub interactions of DUBs and the NF-κB effectors. A20 binds to M1-linked bridges between RIPK1 and NEMO, prohibiting other Ub-binding proteins from attachment and sequestrating the NF-κB-inducing signal. It also removes Ub chains from IκBα, thus disallowing its degradation and it replaces the Ub chain (from K63 to K48) on RIPK1, thus marking it for degradation. In addition, there is CYLD that competes with A20 on NEMO or RIPK1 degradation and affects several signaling components. Lastly, OTUB1 stabilizes p100, a progenitor protein of p52, which inhibits proteolytic degradation of p100 to 52, thus sequestrating the signal in the non-canonical NF-κB pathway. Based on [35][37][39][43]. Legend: black arrow—indicates non-antagonistic interaction or the direction of signal transduction in (A) or only points out the target protein in (B), red arrow—indicates antagonistic or competitive interaction, red arrow with a blunt end—indicates inhibition in (A,B), thunder-like symbol—indicates deubiquitination in (B). A20—tumor necrosis factor, alpha-induced protein 3, cIAP1/2—cellular inhibitor of apoptosis protein-1/2, CYLD—cylindromatosis tumor suppressor protein, IKK1/2—inhibitor of nuclear factor kappa B kinase alpha/beta, IKK1/2—inhibitor of nuclear factor kappa B kinase complex, IκBα—inhibitor of nuclear factor kappa B alpha, LUBAC—linear Ub chain assembly complex, NEMO—inhibitor of nuclear factor kappa-B kinase subunit gamma, NF-κB—nuclear factor kappa-light-chain-enhancer of activated B cells, OTUB1—ubiquitin thioesterase otubain-1, P—an indication of a protein-specific phosphorylation, p50—nuclear factor NF-kappa-B p105 subunit, p52—nuclear factor NF-kappa-B p100 subunit, p65 (RelA)—v-rel avian reticuloendotheliosis viral oncogene homolog A, RelB—v-rel avian reticuloendotheliosis viral oncogene homolog B, RIPK1—receptor-interacting Ser-Thr protein kinase 1, TAB2/3—TGF-beta-activated kinase 1, TAK1—mitogen-activated protein kinase kinase kinase 7, TNFR1—TNF receptor 1, TNFα—tumor necrosis factor α, TRADD—tumor necrosis factor receptor type 1-associated DEATH domain protein, TRAF2/5—TNF receptor-associated factor 2 and 5, Ub—ubiquitin.
References
- Richards, E. Beverages. Science 1890, 396, 127–131.
- Le Daré, B.; Gicquel, T. Therapeutic applications of ethanol: A review. J. Pharm. Pharm. Sci. 2019, 22, 525–535.
- Oscar-Berman, M.; Marinković, K. Alcohol: Effects on neurobehavioral functions and the brain. Neuropsychol. Rev. 2007, 17, 239–257.
- Thompson, T.; Oram, C.; Correll, C.U.; Tsermentseli, S.; Stubbs, B. Analgesic effects of alcohol: A systematic review and meta-analysis of controlled experimental studies in healthy participants. J. Pain. 2017, 5, 499–510.
- Shield, K.D.; Parry, C.; Rehm, J. Chronic diseases and conditions related to alcohol use. Alcohol Res. 2013, 35, 155–173.
- Rehm, J. The risks associated with alcohol use and alcoholism. Alcohol Res. Health 2011, 34, 135–143.
- Frazier, T.H.; Stocker, A.M.; Kershner, N.A.; Marsano, L.S.; McClain, C.J. Treatment of alcoholic liver disease. Ther. Adv. Gastroenterol. 2011, 4, 63–81.
- Kawaratani, H.; Tsujimoto, T.; Douhara, A.; Takaya, H.; Moriya, K.; Namisaki, T.; Noguchi, R.; Yoshiji, H.; Fujimoto, M.; Fukui, H. The effect of inflammatory cytokines in alcoholic liver disease. Mediators Inflamm. 2013.
- Mandrekar, P.; Szabo, G. Signalling pathways in alcohol-induced liver inflammation. J. Hepatol. 2009, 50, 1258–1266.
- Sun, S.C. The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558.
- Donohue, T.M., Jr. Alcohol-induced steatosis in liver cells. World J. Gastroenterol. 2007, 13, 4974–4978.
- Luedde, T.; Schwabe, R.F. NF-κB in the liver-linking injury, fibrosis and hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 108–118.
- D’Andrea, A.; Pellman, D. Deubiquitinating enzymes: A new class of biological regulators. Crit. Rev. Biochem. Mol. Biol. 1998, 33, 337–352.
- Reyes-Turcu, F.E.; Ventii, K.H.; Wilkinson, K.D. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu. Rev. Biochem. 2009, 78, 363–397.
- Amerik, A.Y.; Hochstrasser, M. Mechanism and function of deubiquitinating enzymes. Biochim. Biophys. Acta Mol. Cell Res. 2004, 1695, 189–207.
- Dixit, V.M.; Green, S.; Sarma, V.; Holzman, L.B.; Wolf, F.W.; O’Rourke, K.; Ward, P.A.; Prochownik, E.V.; Marks, R.M. Tumor necrosis factor-alpha induction of novel gene products in human endothelial cells including a macrophage-specific chemotaxin. J. Biol. Chem. 1990, 265, 2973–2978.
- Da Silva, C.G.; Cervantes, J.R.; Studer, P.; Ferran, C. A20-an omnipotent protein in the liver: Prometheus myth resolved? Mult. Ther. Targets A20 2014, 117–139.
- Catrysse, L.; Fukaya, M.; Sze, M.; Meyerovich, K.; Beyaert, R.; Cardozo, A.K.; Van Loo, G. A20 deficiency sensitizes pancreatic beta cells to cytokine-induced apoptosis in vitro but does not influence type 1 diabetes development in vivo. Cell Death Dis. 2015.
- Catrysse, L.; Farhang Ghahremani, M.; Vereecke, L.; Youssef, S.A.; McGuire, C.; Sze MWeber, A.; Heikenwalder, M.; de Bruin, A.; Beyaert, R.; van Loo, G. A20 prevents chronic liver inflammation and cancer by protecting hepatocytes from death. Cell Death Dis. 2016, 7, e2250.
- Catrysse, L.; Vereecke, L.; Beyaert, R.; van Loo, G. A20 in inflammation and autoimmunity. Trends Immunol. 2014, 1, 22–31.
- Wertz, I.; O’Rourke, K.; Zhou, H.; Eby, M.; Aravind, L.; Seshagiri, S.; Wu, P.; Wiesmann, C.; Baker, R.; Boone, D.L.; et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-κB signalling. Nature 2004, 430, 694–699.
- De, A.; Dainichi, T.; Rathinam, C.V.; Ghosh, S. The deubiquitinase activity of A20 is dispensable for NF-κB signaling. EMBO Rep. 2014, 15, 775–783.
- Lee, E.G.; Boone, D.L.; Chai, S.; Libby, S.L.; Chien, M.; Lodolce, J.P.; Ma, A. Failure to regulate TNF-induced NF-κB and cell death responses in A20-deficient mice. Science 2000, 289, 2350–2354.
- Martens, A.; Priem, D.; Hoste, E.; Vetters, J.; Rennen, S.; Catrysse, L.; Voet, S.; Deelen, L.; Sze, M.; Vikkula, H.; et al. Two distinct ubiquitin-binding motifs in A20 mediate its anti-inflammatory and cell-protective activities. Nat. Immunol. 2020, 21, 381–387.
- Razani, B.; Whang, M.I.; Kim, F.S.; Nakamura, M.C.; Sun, X.; Advincula, R.; Turnbaugh, J.A.; Pendse, M.; Tanbun, P.; Achacoso, P.; et al. Non-catalytic ubiquitin binding by A20 prevents psoriatic arthritis-like disease and inflammation. Nat. Immunol. 2020, 21, 422–433.
- Nakagawa, M.M.; Thummar, K.; Mandelbaum, J.; Pasqualucci, L.; Rathinam, C.V. Lack of the ubiquitin-editing enzyme A20 results in loss of hematopoietic stem cell quiescence. J. Exp. Med. 2015, 212, 203–216.
- Feng, Y.; Zhang, Y.; Cai, Y.; Liu, R.; Lu, M.; Li, T.; Fu, Y.; Guo, M.; Huang, H.; Ou, Y.; et al. A20 targets PFKL and glycolysis to inhibit the progression of hepatocellular carcinoma. Cell Death Dis. 2020, 11, 89.
- Chen, H.; Hu, L.; Luo, Z.; Zhang, J.; Zhang, C.; Qiu, B.; Dong, L.; Tan, Y.; Ding, J.; Tang, S.; et al. A20 suppresses hepatocellular carcinoma proliferation and metastasis through inhibition of Twist1 expression. Mol. Cancer 2015, 14.
- Balakirev, M.Y.; Tcherniuk, S.O.; Jaquinod, M.; Chroboczek, J. Otubains: A new family of cysteine proteases in the ubiquitin pathway. EMBO Rep. 2003, 4, 517–522.
- Wiener, R.; DiBello, A.T.; Lombardi, P.M.; Guzzo, C.M.; Zhang, X.; Matunis, M.J.; Wolberger, C. E2 ubiquitin-conjugating enzymes regulate the deubiquitinating activity of OTUB1. Nat. Struct. Mol. Biol. 2013, 20, 1033–1039.
- Chroma, K.; Mistrik, M.; Moudry, P.; Gursky, J.; Liptay, M.; Strauss, R.; Skrott, Z.; Vrtel, R.; Bartkova, J.; Kramara, J.; et al. Tumors overexpressing RNF168 show altered DNA repair and responses to genotoxic treatments, genomic instability and resistance to proteotoxic stress. Oncogene 2017, 36, 2405–2422.
- Nakada, S.; Tai, I.; Panier, S.; Al-Hakim, A.; Iemura, S.; Juang, Y.C.; O’Donnell, L.; Kumakubo, A.; Munro, M.; Sicheri, F.; et al. Non-canonical inhibition of DNA damage-dependent ubiquitination by OTUB1. Nature 2010, 466, 941–946.
- Wiener, R.; Zhang, X.; Wang, T.; Wolberger, C. The mechanism of OTUB1-mediated inhibition of ubiquitination. Nature 2012, 483, 618–622.
- Mulas, F.; Wang, X.; Song, S.; Nishanth, G.; Yi, W.; Brunn, A.; Larsen, P.K.; Isermann, B.; Kalinke, U.; Barragan, A.; et al. The deubiquitinase OTUB1 augments NF-κB-dependent immune responses in dendritic cells in infection and inflammation by stabilizing UBC13. Cell. Mol. Immunol. 2020.
- Li, Y.; Yang, J.Y.; Xie, X.; Jie, Z.; Zhang, L.; Shi, J.; Lin, D.; Gu, M.; Zhou, X.; Li, H.S.; et al. Preventing abnormal NF-κB activation and autoimmunity by Otub1-mediated p100 stabilization. Cell Res. 2019, 29, 474–485.
- Sun, S.C. CYLD: A tumor suppressor deubiquitinase regulating NF-kappaB activation and diverse biological processes. Cell Death Differ. 2010, 17, 25–34.
- Lork, M.; Verhelst, K.; Beyaert, R. CYLD, A20 and OTULIN deubiquitinases in NF-κB signaling and cell death: So similar, yet so different. Cell Death Differ. 2017, 24, 1172–1183.
- Wooten, M.W.; Geetha, T.; Babu, J.R.; Seibenhener, M.L.; Peng, J.; Cox, N.; Diaz-Meco, M.T.; Moscat, J. Essential role of sequestosome 1/p62 in regulating accumulation of Lys63-ubiquitinated proteins. J. Biol. Chem. 2008, 283, 6783–6789.
- Harhaj, E.W.; Dixit, V.M. Deubiquitinases in the regulation of NF-κB signaling. Cell Res. 2011, 21, 22–39.
- Harhaj, E.W.; Dixit, V.M. Regulation of NF-κB by deubiquitinases. Immunol. Rev. 2012, 246, 107–124.
- Damgaard, R.B.; Jolin, H.E.; Allison, M.E.D.; Davies, S.E.; Titheradge, H.L.; McKenzie, A.N.J.; Komander, D. OTULIN protects the liver against cell death, inflammation, fibrosis, and cancer. Cell Death Differ. 2020, 27, 1457–1474.
- Fiil, B.K.; Gyrd-Hansen, M. OTULIN deficiency causes auto-inflammatory syndrome. Cell Res. 2016, 26, 1176–1177.
- Martens, A.; van Loo, G. A20 at the Crossroads of cell death, inflammation, and autoimmunity. Cold Spring Harb. Perspect. Biol. 2020, 12.

