Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Qu, L.; Matz, A.J.; Karlinsey, K.; Cao, Z.; Vella, A.T.; Zhou, B. Macrophage, Meta-Inflammation, and Inflammaging. Encyclopedia. Available online: https://encyclopedia.pub/entry/36402 (accessed on 30 July 2026).
Qu L, Matz AJ, Karlinsey K, Cao Z, Vella AT, Zhou B. Macrophage, Meta-Inflammation, and Inflammaging. Encyclopedia. Available at: https://encyclopedia.pub/entry/36402. Accessed July 30, 2026.
Qu, Lili, Alyssa J. Matz, Keaton Karlinsey, Ziming Cao, Anthony T. Vella, Beiyan Zhou. "Macrophage, Meta-Inflammation, and Inflammaging" Encyclopedia, https://encyclopedia.pub/entry/36402 (accessed July 30, 2026).
Qu, L., Matz, A.J., Karlinsey, K., Cao, Z., Vella, A.T., & Zhou, B. (2022, November 24). Macrophage, Meta-Inflammation, and Inflammaging. In Encyclopedia. https://encyclopedia.pub/entry/36402
Qu, Lili, et al. "Macrophage, Meta-Inflammation, and Inflammaging." Encyclopedia. Web. 24 November, 2022.
Copy Citation
Macrophages are central players in systemic inflammation associated with obesity and aging, termed meta-inflammation and inflammaging. Activities of macrophages elicited by the two chronic conditions display shared and distinct patterns mechanistically, resulting in multifaceted actions for their pathogenic roles. Drastically expanded tissue macrophage populations under obesity and aging stress attribute to both enhanced recruitment and local expansion.
Macrophage
Obesity
Aging
Meta-inflammation
Inflammaging
1. Introduction
Aging is a progressive, multifactorial process, characterized by functional deterioration at the organ, tissue, cellular, and molecular levels. A state of chronic, low-grade systemic inflammation during aging is termed inflammaging, and is associated with aging-induced health risk [1]. During the last few decades, mounting evidence suggests a central role for macrophage malfunction in inflammaging development.
In addition to the expanding elderly population, the prevalence of obesity is rising. Obesity is the fifth leading cause of death worldwide and by 2030, 57.8% of the global adult population is projected to be overweight or obese [2]. Obesity-driven metabolic dysfunction and chronic inflammation, termed meta-inflammation, can progress toward serious conditions such as type II diabetes mellitus (T2D), non-alcoholic fatty liver disease (NAFLD) [3], cardiovascular disease, stroke, and cancers [4][5][6][7]. Studies revealed that higher BMI during young adulthood and middle age significantly increases the chance of hospitalization and mortality after 65 years of age [8]. Fat accumulation in the trunk and visceral areas, which is common in elderly adults, displays a greater health hazard than on hips and limbs, attributed to enhanced inflammatory features associated with these fat depots and phenotypically resemble cellular signatures revealed in obesity [8][9][10]. Further investigation into the association between meta-inflammation and inflammaging is necessary to improve mitigation strategies for age- and obesity-related health risks.
Macrophages are the most common immune population in almost all tissues and act as major players to ensure the homeostatic function of their host tissues under normal physiological conditions. Meanwhile, the plasticity of macrophages also allows them to respond to acute or chronic cues with swift, fine-tuned, and diverse responses, executing crucial functions during pathogen invasion or modulating chronic stress, such as in obesity and aging [11]. Obese and elderly individuals both have higher levels of circulating inflammatory markers, indicating chronic inflammation [12]. Macrophages, as primary mediators of inflammation in the circulation and tissues, act as a “bridge” to connect obesity and aging.
2. Macrophage, Meta-Inflammation, and Inflammaging
2.1. Origin and Distribution of Macrophages
Macrophages are crucial players in innate immunity and the most widespread immune lineage in almost all tissues. They derive from either embryonic precursors or bone marrow (BM) hematopoietic stem/progenitor cells. Embryo-derived macrophages are crucial for proper tissue development and remodeling during fetal development [5]. Disruption of these populations results in growth retardation or mortality [13]. Tissue-resident macrophages, including liver Kupffer cells, skin Langerhans cells, brain microglia, lung alveolar macrophages, and others, were revealed to be of embryonic origin through elegant studies employing gate-mapping strategies in genetic mouse models. Embryo-derived macrophage populations maintain their populations in a self-sustaining manner under normal conditions [13][14][15]. In adulthood, macrophages evolve from BM-derived progenitors to maintain circulating monocyte/macrophage populations and respond to acute or chronic demands from tissues. Upon injury, infection, or sterile inflammation, monocytes are recruited and terminally differentiate into macrophages, as shown in Figure 1.
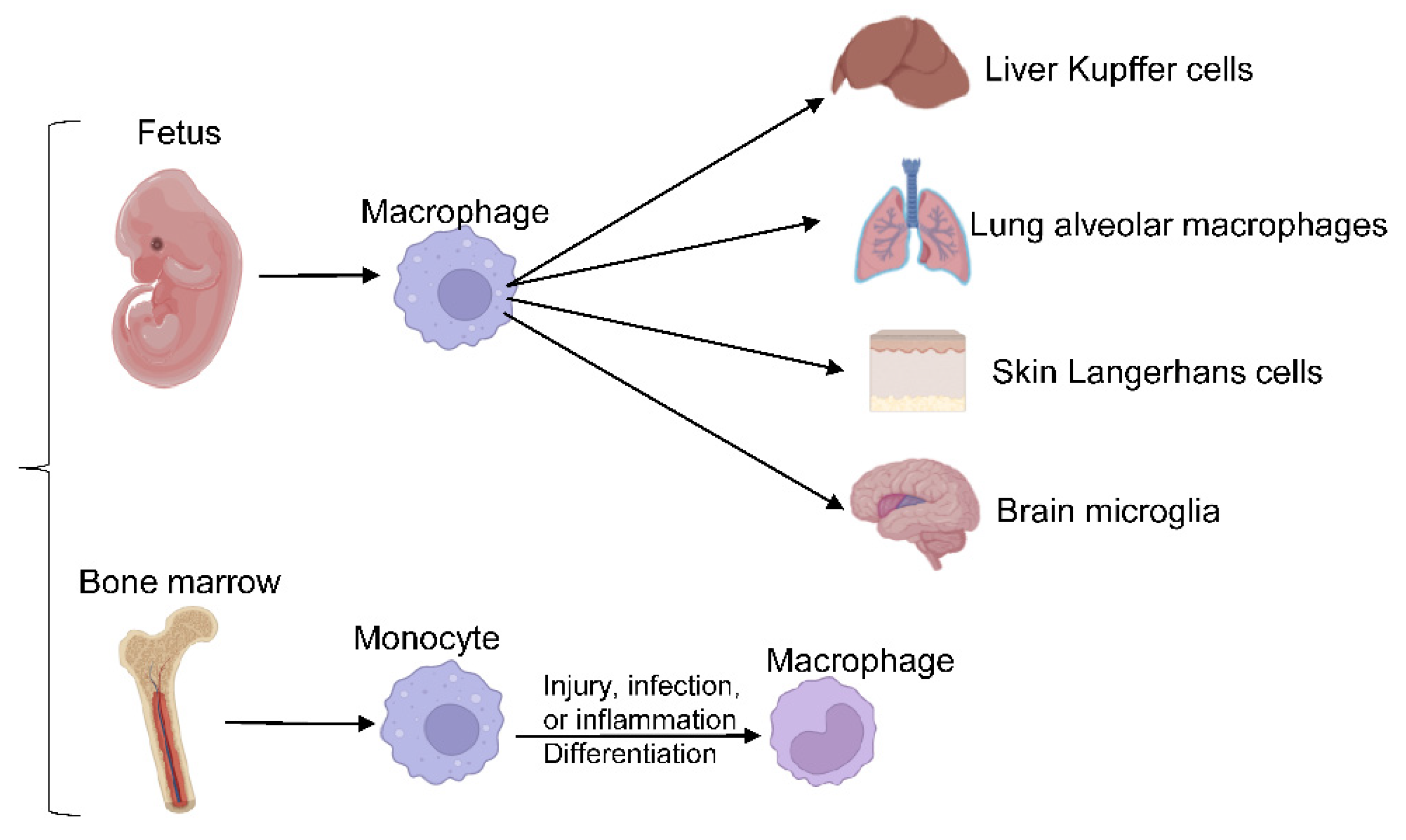
Figure 1. Macrophages differentiation. Embryo-derived macrophages differentiate into different tissue-resident macrophages, responding to acute or chronic demands from tissues in adulthood.
Both BM-derived and embryo-derived macrophages commonly coexist in the same tissues and work together to accomplish their functions. Various types of macrophages with unique functions and phenotypes are assembled in the gut in order to deal with the normal gut flora and oral-derived antigens [16]. Secondary lymphoid organs are also populated by various types of macrophages, such as marginal zone macrophages and sub-capsular sinus macrophages. Unique macrophages accumulate in the immune-privileged tissues of the brain, eye, and testes to perform pivotal roles in tissue remodeling and homeostasis [6][17].
2.2. Pathogenic Changes of Macrophages during Meta-Inflammation or Inflammaging and Related Molecular Mechanism
Depending on the stimuli or signals from the microenvironment, macrophages can quickly employ highly orchestrated signaling networks to mount appropriate responses.
2.2.1. The Plasticity of Macrophage Activation and Relevant Molecular Mechanism
Macrophages are highly plastic to allow fast phenotypic switches for rapid responses to complex and diverse microenvironmental cues. Several models have been proposed to depict macrophage-activation features. The most popular model, macrophage polarization, characterizes macrophages into two classes: classically activated (M1) and alternatively activated (M2) macrophages [18]. This model is based on ex vivo observations of macrophage responses to T helper 1 (Th1) or Th2 stimuli and has become antiquated. Modifications to this model have sought to capture macrophages’ sophisticated actions in various tissues and physiological contexts [11][17][19]. However, no currently available model allows for comprehensive annotation of complex macrophage features under different conditions [17][20]. Researchers created a high-resolution macrophage annotation program termed MacSpectrum [11]. MacSpectrum annotates macrophages based on their differentiation and polarization states, using two indices to capture dynamic transitions of macrophage actions under both in vitro and in vivo conditions.
Pro-inflammatory markers are highly expressed in many elderly or obese adults, even in the absence of clinically active disease [21]. For instance, macrophages will detect and respond to these pro-inflammatory mediators in both aging and obese tissues. Increased chemokine and cytokine levels were reported in the elderly and obese blood with overlapping patterns [8]. Supporting this theory, the adipose tissue macrophage (ATM) compartment in old mice is drastically different from that in young mice, with a higher percentage of M1-like, pro-inflammatory macrophages than their young counterparts, without an increase in overall macrophage number (unlike in obesity) [22]. Of note, studies using aging mice or humans are confounded by inherent visceral adipose tissue accumulation, as humans, mice, and rats advance with age. Similarly, visceral adipose tissue and systemic inflammation are hallmarks of obesity in both mouse models and human studies, characterized by increased number of macrophages, and other immune cells attribute to residential macrophage expansion and increased recruitment [8]. Moreover, the proportion of inflammation status of ATMs were drastically increased, resulting in higher levels of proinflammatory cytokine production, and systemic low-degree inflammation [23].
The mechanisms underlying macrophage polarized activation under meta-inflammation or inflammaging have been extensively investigated. Under the stress of obesity or aging, adipose tissue (AT) secretes certain adipokines/chemokines, which drive the recruitment of circulating immune cells. In addition, anti-inflammatory AT-derived adipokine/cytokine production is reduced, further exacerbating the overall AT inflammatory profile. One such anti-inflammatory adipokine is adiponectin, which is reduced in obese AT and plasma. In culture, human monocytes treated with recombinant adiponectin differentiate to an anti-inflammatory, M2-like phenotype [24][25][26]. Other than adipokine dysfunction, DAMPs and PAMPs, including LPS, interferon-γ (IFN-γ), and other TLR activators, are elevated in obese or aging individuals. These stimuli promote the M1 polarization of macrophages and activate downstream adapter proteins (e.g., MyD88), which induce the expression of pro-inflammatory genes, such as IL-1β, IL-18, and TNF-α [27]. In addition, epigenetic modifications exert an additional layer of impact on macrophage activation heterogeneity. Inhibition of histone deacetylases (HDACs) in obesity-induced diabetes models decreased body weight and blood glucose, and increased insulin sensitivity, in part, through suppressing pro-inflammatory macrophage activation and promoting alternative (M2) activation [28][29]. Furthermore, obese adults with T2D have higher levels of plasma IFN-γ, which can selectively silence the anti-inflammatory pathways by recruiting EZH2 and H3K27me3 to anti-inflammatory cytokine gene loci [30]. Interestingly, in a mouse model of aging-induced osteoporosis, similar molecular pattern shift was also observed. It was reported that increased EZH2 and decreased HDAC9 could promote age-associated osteoporosis and likely through increasing macrophage-dependent recruitment of T cells to the joints [31].
2.2.2. Interplays of Macrophages with Other Immune Cells in Meta-Inflammation or Inflammaging
Macrophages, as pivotal to immune response, can interplay with other immune cells during inflammation, especially T cells. One primary aspect of macrophage function is interaction with adaptive immune cells, such as T cells, as antigen-presenting cells (APCs) to engaging adaptive immunity obesity or aging. In obese mice, macrophages with a deficiency of MHC II reduced the accumulation of effector/memory phenotype CD4+ T cells in white adipose tissue (WAT), which indicated the significance of MHCII-dependent signals from adipose tissue macrophages (ATM) in regulating T cell activation and maturation in meta-information [32]. In parallel, under inflammaging conditions such as Rheumatoid arthritis (RA), macrophages can directly regulate T cells recruitment, differentiation, and activation into RA synovium [33]. In the RA mouse model, increased infiltration of macrophages in RA synovium further attracted CXCR6+ T to the site to improve synovial inflammation [33][34]. Besides recruitment, macrophages can also promote CD4 T helper cells to differentiate to be T helper cells in RA mouse models, which was confirmed in human studies [35][36]. Moreover, macrophages can polarize CD4+ T or Th17 cells through secretion of IL-12, IL-1β, or IL-6 in RA [34][37]. It is necessary to further mechanistically investigate the interplays of macrophage and adaptive immune cells and their pathological impact on health risks associated with obesity and aging.
2.2.3. Cellular Metabolism Reprogramming and Associated Mechanism
Metabolic reprogramming of macrophages is vital to developing and maintaining their functional phenotypes. Under meta-inflammation and inflammaging conditions, macrophages preferentially polarize towards a pro-inflammatory state (M1-like). M2-like macrophages are more abundant in young, lean individuals. Accordingly, macrophage intracellular metabolism is also reprogrammed to support such activation demands, as shown in Figure 2.
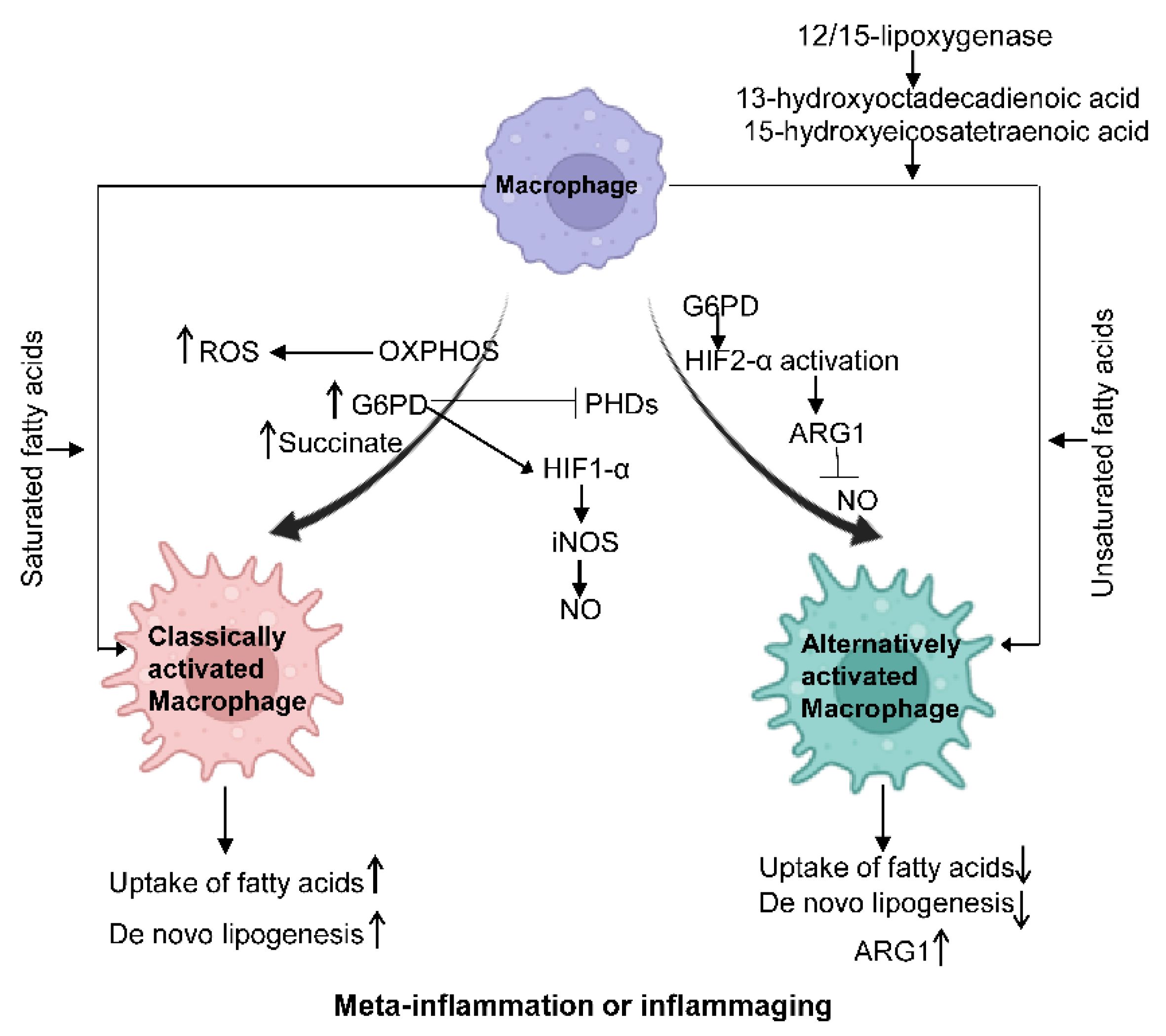
Figure 2. Macrophage metabolism changes under different states. Classically activated.
Macrophages show increased glycolysis, ROS, G6PD, and the TCA intermediate succinate. PHD expression is inhibited, and HIF1-α is activated, which further promotes the production of NO. In alternatively activated macrophages, HIF2-α is activated through ARG1 to suppress NO production. Saturated fatty acids can facilitate the polarization of classically activated macrophages, while unsaturated fatty acids can promote the polarization of alternatively activated macrophages. IL-4-induced 12/15-lipoxygenase can foster alternative macrophage activation via the generation of PPARγ ligands such as 13-hydroxyoctadecadienoic acid and 15-hydroxyeicosatetraenoic acid.
Glucose Metabolism
The rapid activation of macrophages demands a fast energy supply, evidenced by increased rates of glycolysis and mitochondrial oxidative phosphorylation (OXPHOS), a shared metabolic character in both aging and obesity. It should be noted that as the the OXPHOS rate increases, its by-product reactive oxygen species (ROS) increases, a major cell stressor [32]. Hexokinase activity, glucose-6-phosphate dehydrogenase (G6PD), and TCA intermediate succinate are increased in M1-polarized macrophages, which, in turn, inhibit the expression of prolyl hydroxylases (PHDs) and stabilize hypoxia-inducible factor 1-α (HIF1-α) via hydroxylation, resulting in IL-1β production in macrophages [38][39]. On the other side, HIF2-α activation primarily occurs in alternatively activated macrophages and promotes the expression of Arginase1 and suppresses NO production [35]. Further, the mechanistic target of rapamycin complex 1 (mTORC1) modulation impacts metabolic dysfunction. mTORC1 is an integral part of insulin, glucose, leptin, and growth factor signaling cascades to regulate metabolism. Defects in macrophage mTORC1 expression protect against obesity-induced AT inflammation and IR, likely due to a suppressed rate of glycolysis and caspase-1 activity, possibly inhibiting M1 polarization [40]. Additionally, suppressed mTORC1 can inhibit M1 polarization during the aging process [2][3][4][5].
Fatty Acid Metabolism
In macrophages, lipoprotein particles, the primary source of fatty acids, are crucial for polarized macrophage activation [37][38][39]. M1 macrophages have reduced uptake of fatty acids compared to M2 macrophages. In obesity, PPARγ ligands, such as 13-hydroxyoctadecadienoic acid and 15-hydroxyeicosatetraenoic acid, can be generated by the IL-4 inducible 12/15-lipoxygenase to foster alternative macrophage activation. In healthy adults aged over 50 years, the fatty acid profile changes following aging sabotage the switch from M1 to M2 through suppressing PPARγ activity [41]. Saturated fatty acids activate NLRP3 in primed macrophages, while unsaturated fatty acids suppress inflammasome activation [37][38][39]. Lastly, triglycerides taken up by macrophages can undergo lipolysis to generate fatty acids to support M2 activation, which is an example of cell-intrinsic lysosomal lipolysis supporting macrophage alternative function [40].
Amino Acid Metabolism
The role of some amino acids, including glutamine and arginine, in macrophage polarization has been extensively investigated; L-arginine metabolism is strictly controlled in macrophages through iNOS and ARG1. L-arginine can be broken down to NO and L-citrulline by iNOS or to ornithine and urea by ARG1. The expression of iNOS and the production of NO are important features of inflammatory activation of macrophages in both mouse and human studies [42][43]. Additionally, alternative activation of macrophages is characterized by highly expressed ARG1 that promotes polyamine generation, which is essential for collagen synthesis and cell proliferation [44][45].
Other Metabolic Changes
In addition to glucose, fatty acids, and amino acid metabolism, macrophage vitamins and iron metabolism are also altered in elderly obese individuals. Vitamin D shortage can trigger inflammation and has been linked to immune-mediated diseases such as T2D [46]. Vitamin A regulates the differentiation and function of macrophages and enhances their phagocytic capacity in mouse studies [45]. For iron metabolism reported in mouse models, inflammatory macrophages employ an iron retention program and display increased Ferritin and decreased Ferroportin phenotype; in contrast, anti-inflammatory macrophages are marked with the opposite phenotype and favor an iron-export mode [47].
References
- Crunkhorn, S. Reversing inflammaging. Nat. Rev. Drug Discov. 2020, 19, 168.
- GBD 2017 Risk Factor Collaborators. Global, regional, and national comparative risk assessment of 84 behavioural, environmental and occupational, and metabolic risks or clusters of risks for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1923–1994.
- Qu, L.L.; Yu, B.; Li, Z.; Jiang, W.X.; Jiang, J.D.; Kong, W.J. Gastrodin Ameliorates Oxidative Stress and Proinflammatory Response in Nonalcoholic Fatty Liver Disease through the AMPK/Nrf2 Pathway. Phytother. Res. 2016, 30, 402–411.
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462.
- Varol, C.; Mildner, A.; Jung, S. Macrophages: Development and tissue specialization. Annu. Rev. Immunol. 2015, 33, 643–675.
- Ginhoux, F.; Jung, S. Monocytes and macrophages: Developmental pathways and tissue homeostasis. Nat. Rev. Immunol. 2014, 14, 392–404.
- Li, C.; Xu, M.M.; Wang, K.; Adler, A.J.; Vella, A.T.; Zhou, B. Macrophage polarization and meta-inflammation. Transl. Res. 2018, 191, 29–44.
- Reyes-Farias, M.; Fos-Domenech, J.; Serra, D.; Herrero, L.; Sanchez-Infantes, D. White adipose tissue dysfunction in obesity and aging. Biochem. Pharmacol. 2021, 192, 114723.
- Yan, L.L.; Daviglus, M.L.; Liu, K.; Stamler, J.; Wang, R.; Pirzada, A.; Garside, D.B.; Dyer, A.R.; Van Horn, L.; Liao, Y.; et al. Midlife body mass index and hospitalization and mortality in older age. JAMA 2006, 295, 190–198.
- Mouton, A.J.; Li, X.; Hall, M.E.; Hall, J.E. Obesity, Hypertension, and Cardiac Dysfunction: Novel Roles of Immunometabolism in Macrophage Activation and Inflammation. Circ. Res. 2020, 126, 789–806.
- Li, C.; Menoret, A.; Farragher, C.; Ouyang, Z.; Bonin, C.; Holvoet, P.; Vella, A.T.; Zhou, B. Single cell transcriptomics based-MacSpectrum reveals novel macrophage activation signatures in diseases. JCI Insight 2019, 5, e126453.
- Oishi, Y.; Manabe, I. Macrophages in age-related chronic inflammatory diseases. NPJ Aging Mech. Dis. 2016, 2, 16018.
- Yona, S.; Kim, K.W.; Wolf, Y.; Mildner, A.; Varol, D.; Breker, M.; Strauss-Ayali, D.; Viukov, S.; Guilliams, M.; Misharin, A.; et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 2013, 38, 79–91.
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 2013, 38, 792–804.
- Schulz, C.; Gomez Perdiguero, E.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.; Pollard, J.W.; et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 2012, 336, 86–90.
- Davis, M.J.; Tsang, T.M.; Qiu, Y.; Dayrit, J.K.; Freij, J.B.; Huffnagle, G.B.; Olszewski, M.A. Macrophage M1/M2 polarization dynamically adapts to changes in cytokine microenvironments in Cryptococcus neoformans infection. mBio 2013, 4, e00264-00213.
- Das, A.; Sinha, M.; Datta, S.; Abas, M.; Chaffee, S.; Sen, C.K.; Roy, S. Monocyte and macrophage plasticity in tissue repair and regeneration. Am. J. Pathol. 2015, 185, 2596–2606.
- Tilg, H.; Hotamisligil, G.S. Nonalcoholic fatty liver disease: Cytokine-adipokine interplay and regulation of insulin resistance. Gastroenterology 2006, 131, 934–945.
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20.
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896.
- Ferrucci, L.; Semba, R.D.; Guralnik, J.M.; Ershler, W.B.; Bandinelli, S.; Patel, K.V.; Sun, K.; Woodman, R.C.; Andrews, N.C.; Cotter, R.J.; et al. Proinflammatory state, hepcidin, and anemia in older persons. Blood 2010, 115, 3810–3816.
- Lumeng, C.N.; Liu, J.; Geletka, L.; Delaney, C.; Delproposto, J.; Desai, A.; Oatmen, K.; Martinez-Santibanez, G.; Julius, A.; Garg, S.; et al. Aging is associated with an increase in T cells and inflammatory macrophages in visceral adipose tissue. J. Immunol. 2011, 187, 6208–6216.
- Lumeng, C.N.; DelProposto, J.B.; Westcott, D.J.; Saltiel, A.R. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes 2008, 57, 3239–3246.
- Ohashi, K.; Parker, J.L.; Ouchi, N.; Higuchi, A.; Vita, J.A.; Gokce, N.; Pedersen, A.A.; Kalthoff, C.; Tullin, S.; Sams, A.; et al. Adiponectin promotes macrophage polarization toward an anti-inflammatory phenotype. J. Biol. Chem. 2010, 285, 6153–6160.
- Lovren, F.; Pan, Y.; Quan, A.; Szmitko, P.E.; Singh, K.K.; Shukla, P.C.; Gupta, M.; Chan, L.; Al-Omran, M.; Teoh, H.; et al. Adiponectin primes human monocytes into alternative anti-inflammatory M2 macrophages. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H656–H663.
- Mandal, P.; Park, P.H.; McMullen, M.R.; Pratt, B.T.; Nagy, L.E. The anti-inflammatory effects of adiponectin are mediated via a heme oxygenase-1-dependent pathway in rat Kupffer cells. Hepatology 2010, 51, 1420–1429.
- Madore, C.; Yin, Z.; Leibowitz, J.; Butovsky, O. Microglia, Lifestyle Stress, and Neurodegeneration. Immunity 2020, 52, 222–240.
- Brown, J.D.; Lin, C.Y.; Duan, Q.; Griffin, G.; Federation, A.; Paranal, R.M.; Bair, S.; Newton, G.; Lichtman, A.; Kung, A.; et al. NF-kappaB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol. Cell 2014, 56, 219–231.
- Foster, S.L.; Hargreaves, D.C.; Medzhitov, R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature 2007, 447, 972–978.
- Kang, K.; Park, S.H.; Chen, J.; Qiao, Y.; Giannopoulou, E.; Berg, K.; Hanidu, A.; Li, J.; Nabozny, G.; Kang, K.; et al. Interferon-gamma Represses M2 Gene Expression in Human Macrophages by Disassembling Enhancers Bound by the Transcription Factor MAF. Immunity 2017, 47, 235–250.e234.
- Chen, Y.H.; Chung, C.C.; Liu, Y.C.; Yeh, S.P.; Hsu, J.L.; Hung, M.C.; Su, H.L.; Li, L.Y. Enhancer of Zeste Homolog 2 and Histone Deacetylase 9c Regulate Age-Dependent Mesenchymal Stem Cell Differentiation into Osteoblasts and Adipocytes. Stem Cells 2016, 34, 2183–2193.
- Cho, K.W.; Morris, D.L.; DelProposto, J.L.; Geletka, L.; Zamarron, B.; Martinez-Santibanez, G.; Meyer, K.A.; Singer, K.; O’Rourke, R.W.; Lumeng, C.N. An MHC II-dependent activation loop between adipose tissue macrophages and CD4+ T cells controls obesity-induced inflammation. Cell Rep. 2014, 9, 605–617.
- Kim, C.H.; Rott, L.; Kunkel, E.J.; Genovese, M.C.; Andrew, D.P.; Wu, L.; Butcher, E.C. Rules of chemokine receptor association with T cell polarization in vivo. J. Clin. Investig. 2001, 108, 1331–1339.
- Stamp, L.K.; Easson, A.; Pettersson, L.; Highton, J.; Hessian, P.A. Monocyte derived interleukin (IL)-23 is an important determinant of synovial IL-17A expression in rheumatoid arthritis. J. Rheumatol. 2009, 36, 2403–2408.
- Evans, H.G.; Gullick, N.J.; Kelly, S.; Pitzalis, C.; Lord, G.M.; Kirkham, B.W.; Taams, L.S. In vivo activated monocytes from the site of inflammation in humans specifically promote Th17 responses. Proc. Natl. Acad. Sci. USA 2009, 106, 6232–6237.
- Yoon, B.R.; Yoo, S.J.; Choi, Y.; Chung, Y.H.; Kim, J.; Yoo, I.S.; Kang, S.W.; Lee, W.W. Functional phenotype of synovial monocytes modulating inflammatory T-cell responses in rheumatoid arthritis (RA). PLoS ONE 2014, 9, e109775.
- Pene, J.; Chevalier, S.; Preisser, L.; Venereau, E.; Guilleux, M.H.; Ghannam, S.; Moles, J.P.; Danger, Y.; Ravon, E.; Lesaux, S.; et al. Chronically inflamed human tissues are infiltrated by highly differentiated Th17 lymphocytes. J. Immunol. 2008, 180, 7423–7430.
- Newsholme, P.; Curi, R.; Gordon, S.; Newsholme, E.A. Metabolism of glucose, glutamine, long-chain fatty acids and ketone bodies by murine macrophages. Biochem. J. 1986, 239, 121–125.
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 2013, 496, 238–242.
- Jiang, H.; Westerterp, M.; Wang, C.; Zhu, Y.; Ai, D. Macrophage mTORC1 disruption reduces inflammation and insulin resistance in obese mice. Diabetologia 2014, 57, 2393–2404.
- Pararasa, C.; Ikwuobe, J.; Shigdar, S.; Boukouvalas, A.; Nabney, I.T.; Brown, J.E.; Devitt, A.; Bailey, C.J.; Bennett, S.J.; Griffiths, H.R. Age-associated changes in long-chain fatty acid profile during healthy aging promote pro-inflammatory monocyte polarization via PPARgamma. Aging Cell 2016, 15, 128–139.
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084.
- Sun, C.; Sun, L.; Ma, H.; Peng, J.; Zhen, Y.; Duan, K.; Liu, G.; Ding, W.; Zhao, Y. The phenotype and functional alterations of macrophages in mice with hyperglycemia for long term. J. Cell Physiol. 2012, 227, 1670–1679.
- Fuentes, E.; Fuentes, F.; Vilahur, G.; Badimon, L.; Palomo, I. Mechanisms of chronic state of inflammation as mediators that link obese adipose tissue and metabolic syndrome. Mediat. Inflamm. 2013, 2013, 136584.
- Van Dyken, S.J.; Locksley, R.M. Interleukin-4- and interleukin-13-mediated alternatively activated macrophages: Roles in homeostasis and disease. Annu. Rev. Immunol. 2013, 31, 317–343.
- Illescas-Montes, R.; Melguizo-Rodriguez, L.; Ruiz, C.; Costela-Ruiz, V.J. Vitamin D and autoimmune diseases. Life Sci. 2019, 233, 116744.
- Winn, N.C.; Volk, K.M.; Hasty, A.H. Regulation of tissue iron homeostasis: The macrophage “ferrostat”. JCI Insight 2020, 5, e132964.
More
Information
Subjects:
Immunology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.9K
Revisions:
2 times
(View History)
Update Date:
25 Nov 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No