Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Marija Pljesa-Ercegovac | -- | 1743 | 2022-11-24 12:15:28 | | | |
| 2 | Conner Chen | Meta information modification | 1743 | 2022-11-28 09:45:40 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Simic, P.; Pljesa, I.; Nejkovic, L.; Jerotic, D.; Coric, V.; Stulic, J.; Kokosar, N.; Popov, D.; Savic-Radojevic, A.; Pazin, V.; et al. Chemotherapy Resistance of Ovarian Cancer. Encyclopedia. Available online: https://encyclopedia.pub/entry/36378 (accessed on 21 June 2026).
Simic P, Pljesa I, Nejkovic L, Jerotic D, Coric V, Stulic J, et al. Chemotherapy Resistance of Ovarian Cancer. Encyclopedia. Available at: https://encyclopedia.pub/entry/36378. Accessed June 21, 2026.
Simic, Petar, Igor Pljesa, Lazar Nejkovic, Djurdja Jerotic, Vesna Coric, Jelena Stulic, Nenad Kokosar, Dunja Popov, Ana Savic-Radojevic, Vladimir Pazin, et al. "Chemotherapy Resistance of Ovarian Cancer" Encyclopedia, https://encyclopedia.pub/entry/36378 (accessed June 21, 2026).
Simic, P., Pljesa, I., Nejkovic, L., Jerotic, D., Coric, V., Stulic, J., Kokosar, N., Popov, D., Savic-Radojevic, A., Pazin, V., & Pljesa-Ercegovac, M. (2022, November 24). Chemotherapy Resistance of Ovarian Cancer. In Encyclopedia. https://encyclopedia.pub/entry/36378
Simic, Petar, et al. "Chemotherapy Resistance of Ovarian Cancer." Encyclopedia. Web. 24 November, 2022.
Copy Citation
Chemotherapy resistance of ovarian cancer, regarded as the most lethal malignant gynecological disease, can be explained by several mechanisms, including increased activity of efflux transporters leading to decreased intracellular drug accumulation, increased efflux of the therapeutic agents from the cell by multidrug-resistance-associated protein (MRP1), enhanced DNA repair, altered apoptotic pathways, silencing of a number of genes, as well as drug inactivation.
ovarian cancer
chemoresistance
glutathione S transferase P1
1. Introduction
Ovarian cancer (OC) is regarded as the most lethal malignant gynecological disease, with overall survival in the range of 45–50% [1][2]. Ranked as the second most common gynecological cancer, ovarian cancer is heterogeneous in nature and divided into three major histopathological groups, of which the epithelial subgroup comprises approximately 90% of the cases worldwide [3]. According to statistics, it is estimated that annually worldwide, 230,000 women will be diagnosed with this disease, with lethal outcomes in approximately 150,000 [4]. A great deal of research has been performed towards elucidating the malignant and silent nature of ovarian cancer, and both genetic and epigenetic factors are shown to influence the progression of the disease. Indeed, approximately 10–15% of familial OCs are consequential to BRCA1 and BRCA2 gene mutations [5], while the presence of mutation in TP53 tumor-suppressor gene is found in 60–80% of both familial and sporadic OC cases [6]. The leading cause of survival rates below 50% are advanced-stage disease at the time of diagnosis, chemoresistance, and lack of centralized care for patients, especially in developing counties [7][8]. Regarding the time of diagnosis, detection in the early stage is not satisfactory due to the fact that the most-used diagnostic tools for OC screening, such as transvaginal ultrasound and blood test for CA125 tumor marker, are not efficient enough [9]. Moreover, the major challenges associated with the development of a clinically applicable screening strategy are, on one side, the low prevalence of ovarian cancer and, on the other side, the lack of biomarkers with appropriate sensitivity and specificity. When it comes to treatment, fundamental principles are based on radical surgery combined with platinum-based chemotherapy (carboplatin or cisplatin) in combination with taxane (paclitaxel and docetaxel), to which most patients are initially responsive but, due to development of platinum resistance, a relapse occurs in approximately 80% of OC patients [10]. In platinum-based chemotherapy-resistant ovarian carcinoma, the treatment is based on usage of gemcitabine, doxorubicin, and bevacizumab [3]. Overall, primary debulking surgery still represents a cornerstone therapy, whereas the tumor burden after the surgery is considered the most important survival factor [11][12].
In recent years, great strides have been made in clarifying the molecular background of ovarian cancer, as well as for understanding the mechanism of drug resistance in OC patients. Well-conduced clinical trials have paved the way for the introduction of novel target therapy, primarily antiangiogenic agents, followed by inhibitors against poly (ADP-ribose) polymerase (PARP) molecules involved in the DNA damage repair processes [13][14]. Although the advanced level treatment options include targeted therapy, immunotherapy, and hormone therapy, chemotherapy is still considered the most vital part of treatment in metastatic ovarian cancer [15].
2. Chemotherapy Resistance of Ovarian Cancer
Regarded as a complex phenomenon, which leads to the development of tolerance and failure in cellular response to treatment with one or multiple chemotherapeutic agents, drug resistance or chemotherapy resistance represents a great concern in everyday clinical practice [16]. Moreover, apart from developing resistance to the applied chemotherapeutic agent, cancer cells may even develop simultaneous cross-resistance to a wide range of drugs that may even be functionally and structurally unrelated to the applied chemotherapeutics [17].
In general, mechanisms of chemoresistance are stratified into two basic categories, including de novo or intrinsic and acquired or extrinsic chemoresistance [16][18]. As in any other cancer, chemotherapy resistance of ovarian cancer can be explained by several basic mechanisms, including increased activity of efflux transporters, leading to decreased intracellular drug accumulation, increased efflux of the therapeutic agents from the cell by multidrug-resistance-associated protein (MRP1) [19], enhanced DNA repair, altered apoptotic pathways, silencing of a number of genes, as well as increased cellular levels of glutathione (GSH) and glutathione transferases (GSTs), which are involved in drug detoxification processes (platinum agents and taxol) and seem to play a very important role in this phenomenon [3][16][20][21] (Figure 1).
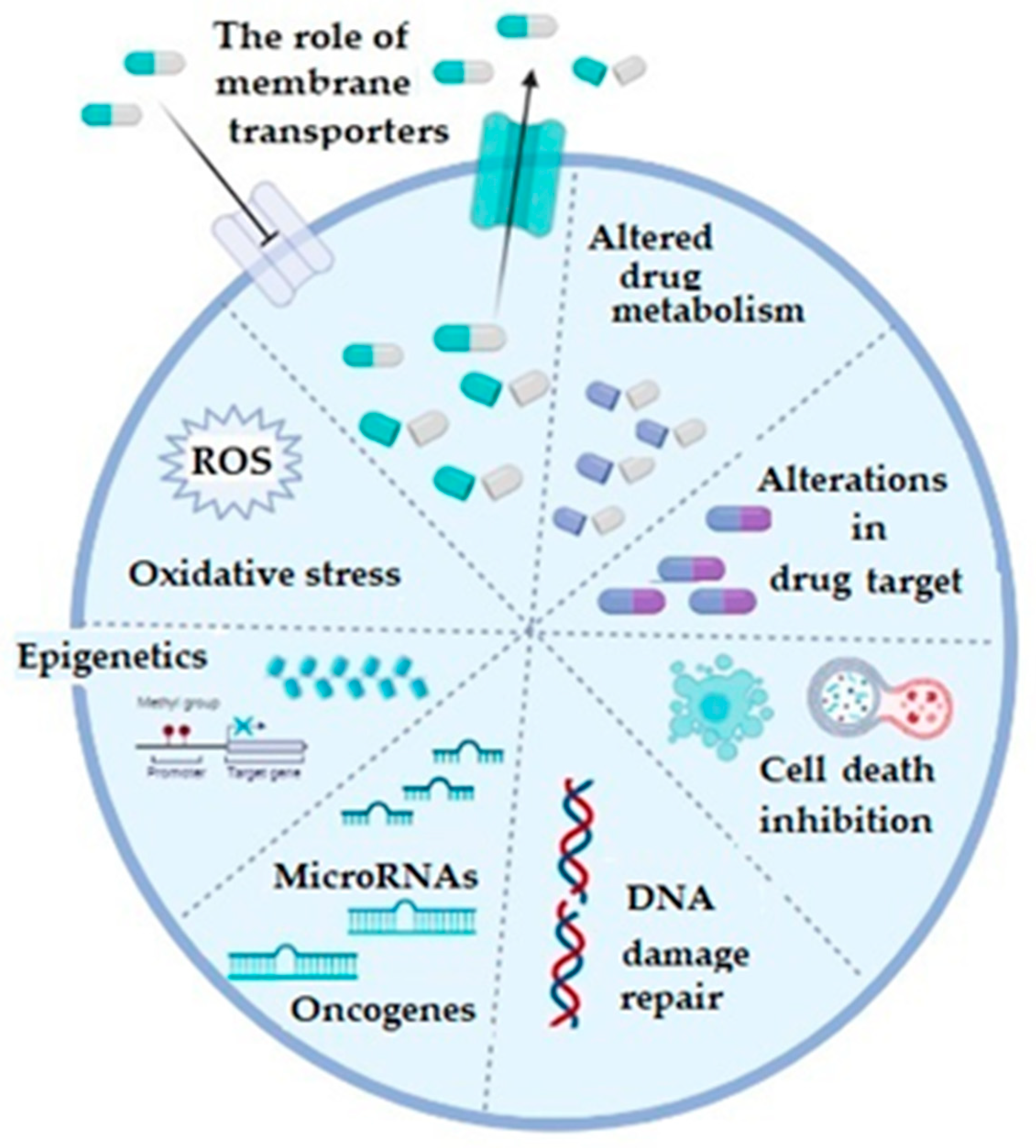
Figure 1. The illustrative summary of cellular mechanisms which are implicated in drug resistance in ovarian cancer; ROS: reactive oxygen species; figure created with Biorender (https://biorender.com/, accessed on 6 November 2022).
Regarding the membrane transporters, both influx and efflux, they participate in the chemoresistance mechanism, which is considered as the most prevalent one and which is based on the reduced cellular accumulation of the applied chemotherapeutic [16][22]. The majority of efflux transporters considered responsible for transporting the drugs outside the ovarian cancer cells, such as doxorubicin, vincristine, cisplatin, paclitaxel, topotecan, and etopiside, belong to a protein superfamily of ATP-binding cassette or ABC transporters [3][23]. Multidrug resistance (MDR)-associated proteins (MRPs), especially the MRP1 (encoded by ABCC1) and MRP2 (encoded by ABCC2) genes, as well as the ATP-dependent glycoprotein P-gp (encoded by ABCB1 gene) and breast cancer resistance protein BCRP (encoded by ABCG2 gene) gained most attention in OC [3]. Increased expression of any of these efflux transporters decreases intracellular concentration of the corresponding drug in OC. Influx or uptake transporters, on the other hand, belong to a wide group of solute carriers (SLC) transporter families, among which organic-anion-transporting proteins (OATPs), especially OATP1B3, is considered important in OC. Precisely, its expression is associated with influx of cis-, cabo-, and oxaliplatin [24]. Additionally, folate receptor α and the reduced folate carrier (RFC) may also be important as differential regulators for the development and progression of ovarian cancer. Namely, in epithelial OC, folate receptor α is highly expressed and increases with the stage of the disease [25][26][27].
Regarding the detoxification pathways of platinum derivatives as well as paclitaxel, as the drugs most commonly used to treat metastatic ovarian cancer patients, glutathione transferases (GSTs) have been recognized as the major enzymes responsible for the conversion of these drugs into less effective forms [28]. Glutathione transferases are a large family of enzymes responsible for catalyzing the conjugation of xenobiotics, including anticancer drugs, with glutathione [29][30]. Great inter-individual differences exist in the GST isoenzyme profile, due to the fact that almost all members of cytosolic GSTs exhibit genetic polymorphism [29][30]. As a consequence, complete lack or alteration in GST enzyme activity might affect the capacity for biotransformation in certain individuals, making them more prone to cancer development. Single nucleotide polymorphisms (SNPs) are mostly responsible for variations identified within genes encoding for cytosolic GSTs and, furthermore, they were associated with numerous diseases, including cancer [29]. In the case of GST pi (GSTP1), SNP leading to amino acid substitution from isoleucine (Ile) to valine (Val) changes catalytic and regulatory properties of the GSTP1 enzyme. Regarding alpha class GST (GSTA1), polymorphism is represented by three, apparently linked, SNPs: −567TOG, −69COT, and −52GOA, which lead to differential expression with lower transcriptional activation of the variant GSTA1*B (−567G, −69T, and −52A) than the common GSTA1*A alleles (−567T, −69C, and −52G). One more SNP, precisely the substitution of Ala to Asp at position 140, changes the deglutathionylase and thioltransferase activity of GST omega class (GSTO1), while, similarly to GSTA1, single nucletide polymorhism (A to G), leading to Asn to Asp substitution at position 142, is related to altered protein levels of GSTO2. On the other hand, deletion polymorphisms of genes encoding for human cytosolic GSTM1 and GSTT1 are rather common in human populations. Approximately half of the population lacks GSTM1 enzyme activity, due to a homozygous deletion of the GSTM1 gene, while in the case of GSTT1, gene homozygous deletion, with consequential lack of GSTT1 enzyme activity, is present in approximately 20% of Caucasians (Table 1) [29][31][32][33][34].
Table 1. Distribution of common GST polymorphisms in humans.
| Gene | rs | Genotype | Distribution in Caucasians (%) |
|---|---|---|---|
| GSTA1 | rs3957357 | GSTA1 AA GSTA1 AB/BB (low-activity) 3 |
38 62 |
| GSTP1 | rs1695 | GSTP1 Ile105Ile GSTP1 Ile105Val/Val105Val (variant) 4 |
45 55 |
| GSTO1 | rs4925 | GSTO1 Ala140Ala GST01 Ala140Asp/Asp140Asp(variant) 4 |
44 56 |
| GSTO2 | rs156697 | GSTO2 Asn142Asn GSTO2 Asn142Asp/Asp142Asp(variant) 4 |
45 55 |
| GSTT1 | deletion | GST1 active 1 GSTT1 null 2 |
70-80 20-30 |
| GSTM1 | deletion | GSTM1 active GSTM1 null |
50 50 |
1 Active, if at least one active allele present; 2 Null, if no active alleles present; 3 Low activity, if at least one B allele present; 4 and Variant, if at least one Val/Asp/Asp allele present.
Apart from platinum derivatives, GSTs are involved in the development of chemoresistance by detoxification of numerous other chemotherapeutics [31][35], including chlorambucil, cyclophosphamide, melphalan, thiotepa, etc., which are recognized as substrates for GSTs and can be directly inactivated through GST-dependent conjugation reactions (Table 2) [31][36].
Table 2. Substrate specificities of cytosolic GSTs regarding different chemotherapeutics.
| GST Class | Alleles | Substrates |
|---|---|---|
| Alpha | GSTA1*A GSTA1*B GSTA2*A GSTA2*B GSTA2*C GSTA2*D GSTA2*E |
melphalan, chlorambucil, thiotepa, BCNU, brostallicin, and busulphan |
| Mu | GSTM1*0 GSTM1*A GSTM1*B GSTM3*A GSTM3*B GSTM4*A GSTM4*B |
brostallicin, BCNU, ethacrynic acid, and thiopurines |
| Pi | GSTP1*A GSTP1*B GSTP1*C GSTP1*D |
cisplatin, brostallicin, chlorambucil, doxorubicin, ethacrynic acid, cyclophosphamide, and thiotepa |
| Theta | GSTT1*0 GSTT1*A GSTT1*B GSTT2*A GSTT2*B |
BCNU |
Furthermore, GSTs may even be responsible for chemoresistance of non-GST substrate drugs by mechanisms such as interaction with efflux transporters or different signaling molecules involved in regulation of apoptosis [31][37][38][39]. This especially refers to pi class GST (GSTP1), since GSTP1 possesses binding activity toward small and macromolecules, acts as a negative regulator of kinase-dependent apoptotic signaling pathways by forming protein–protein complexes with regulatory mitogen-activated kinases such as JNK1 (c-Jun NH2-terminal kinase), and, in addition to its role in detoxification of potential cancerogenic substances, GSTP1 is capable of increasing drug efflux from the cell, thus contributing to chemoresistance (Figure 2) [30][31][40]. Namely, through interaction with MRP-1, GSTP1 exhibits a synergistic effect on chemoresistance development to ethacrynic acid, chlorambucil, vincristine, and etoposide [41]. Other classes of GSTs, primarily GSTA1 and GSTM1, can contribute to chemoresistance mechanisms as well. Due to the structural homology between GSTA1 and GSTP1, GSTA1 may also suppress JNK1 signaling, while this class of GSTs also contributes to chlorambucil chemoresistance [42]. Similarly, through synergism of GSTM1 and MRP-1, cancer cells are protected from vincristine effects [43]. Furthermore, GSTM1 is capable of forming protein–protein interactions with either apoptosis signal—regulating kinase (ASK1) or thioredoxin (Trx)—in that way contributing to cellular redox-sensitive dynamic equilibrium [31]. Interestingly, by forming protein–protein interaction with tumor necrosis factor receptor-associated factor 2 (TRAF2), GSTP1 prevents ASK1:TRAF2 interaction and, consequently, ASK1 activation. Taken together, it might be proposed that overexpressed glutathione transferases via their catalytic, regulatory, and/or synergistic roles participate in several major mechanisms of chemoresistance.
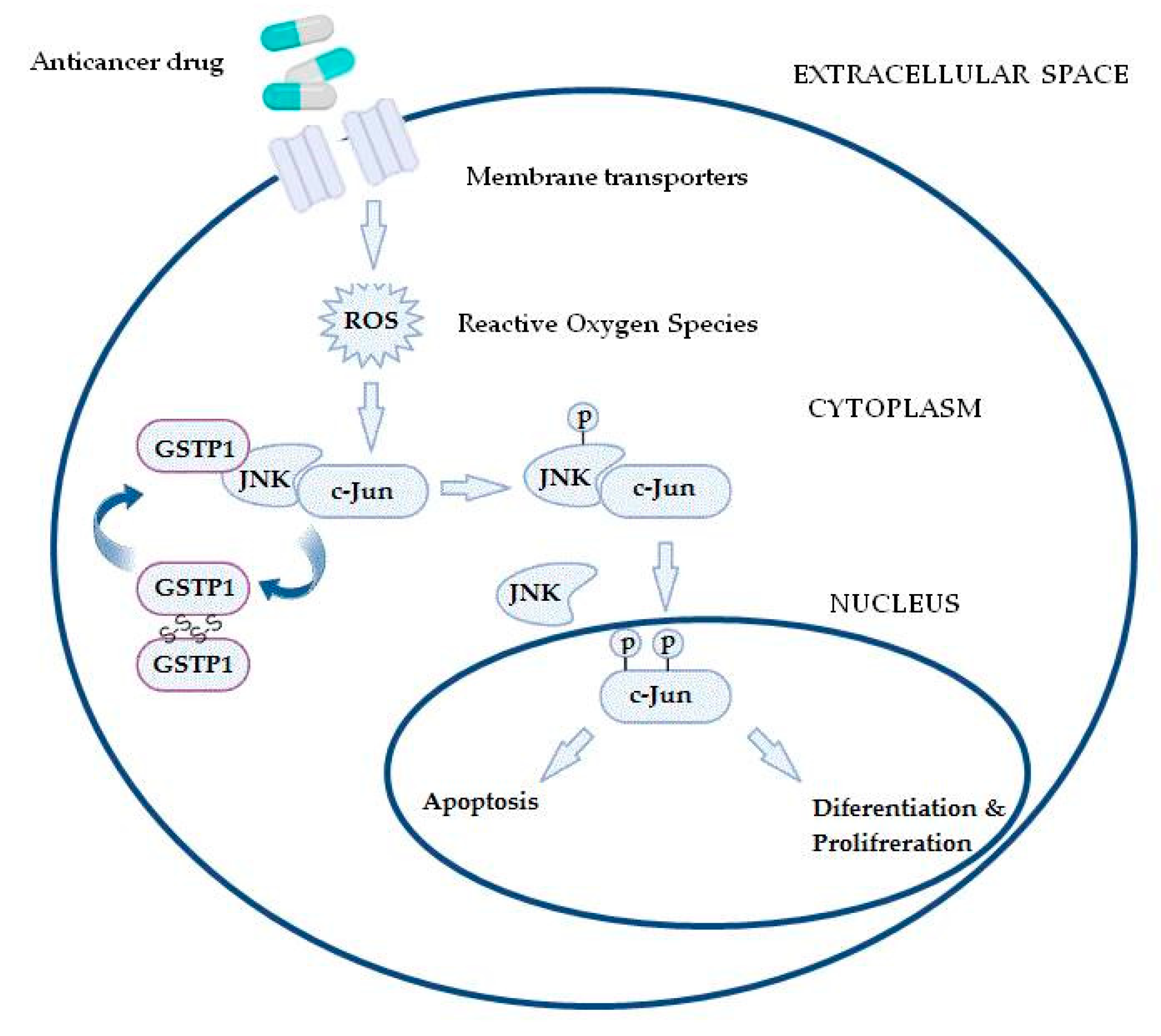
Figure 2. In the case of increased reactive oxygen species (ROS) content, the GSTP1:JNK1 complex dissociates and leads to GSTP1 oligomerisation. Consequently, activated JNK1 causes c-Jun phosphorylation, resulting in its nucleus translocation and initiation of alternative processes, depending on the level of ROS.
References
- Lheureux, S.; Gourley, C.; Vergote, I.; Oza, A.M. Epithelial ovarian cancer. Lancet 2019, 393, 1240–1253.
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33.
- Chandra, A.; Pius, C.; Nabeel, M.; Nair, M.; Vishwanatha, J.K.; Ahmad, S.; Basha, R. Ovarian cancer: Current status and strategies for improving therapeutic outcomes. Cancer Med. 2019, 8, 7018–7031.
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424.
- Neff, R.T.; Senter, L.; Salani, R. BRCA mutation in ovarian cancer: Testing, implications and treatment considerations. Ther. Adv. Med. Oncol. 2017, 9, 519–531.
- Zhang, Y.; Cao, L.; Nguyen, D.; Lu, H. TP53 mutations in epithelial ovarian cancer. Transl. Cancer Res. 2016, 5, 650–663.
- Cliby, W.A.; Powell, M.A.; Al-Hammadi, N.; Chen, L.; Miller, J.P.; Roland, P.Y.; Mutch, D.G.; Bristow, R.E. Ovarian cancer in the United States: Contemporary patterns of care associated with improved survival. Gynecol. Oncol. 2015, 136, 11–17.
- Kim, K.; Hernlund, E.; Hernadi, Z.; Révész, J.; Pete, I.; Szánthó, A.; Bodnar, L.; Madry, R.; Timorek–Lemieszczuk, A.; Bozanovic, T.; et al. Treatment patterns, health care utilization, and costs of ovarian cancer in Central and Eastern Europe using a Delphi panel based on a retrospective chart review. Int. J. Gynecol. Cancer 2013, 23, 823–832.
- Liberto, J.M.; Chen, S.-Y.; Shih, I.-M.; Wang, T.-H.; Wang, T.-L.; Pisanic, T.R. Current and Emerging Methods for Ovarian Cancer Screening and Diagnostics: A Comprehensive Review. Cancers 2022, 14, 2885.
- Damia, G.; Broggini, M. Platinum Resistance in Ovarian Cancer: Role of DNA Repair. Cancers 2019, 11, 119.
- Du Bois, A.; Reuss, A.; Pujade-Lauraine, E.; Harter, P.; Ray-Coquard, I.; Pfisterer, J. Role of surgical outcome as prognostic factor in advanced epithelial ovarian cancer: A combined exploratory analysis of 3 prospectively randomized phase 3 multicenter trials: By the Arbeitsgemeinschaft Gynaekologische Onkologie Studiengruppe Ovarialkarzinom (AGO-OVAR) and the Groupe d’Investigateurs Nationaux Pour les Etudes des Cancers de l’Ovaire (GINECO). Cancer 2009, 115, 1234–1244.
- Kurnit, K.C.; Fleming, G.F.; Lengyel, E. Updates and New Options in Advanced Epithelial Ovarian Cancer Treatment. Obstet. Gynecol. 2021, 137, 108–121.
- Haunschild, C.E.; Tewari, K.S. Bevacizumab use in the frontline, maintenance and recurrent settings for ovarian cancer. Future Oncol. 2020, 16, 225–246.
- Nero, C.; Ciccarone, F.; Pietragalla, A.; Duranti, S.; Daniele, G.; Salutari, V.; Carbone, M.; Scambia, G.; Lorusso, D. Ovarian Cancer Treatments Strategy: Focus on PARP Inhibitors and Immune Check Point Inhibitors. Cancers 2021, 13, 1298.
- Ortiz, M.; Wabel, E.; Mitchell, K.; Horibata, S. Mechanisms of chemotherapy resistance in ovarian cancer. Cancer Drug Resist. 2022, 5, 304–316.
- Norouzi-Barough, L.; Sarookhani, M.R.; Sharifi, M.; Moghbelinejad, S.; Jangjoo, S.; Salehi, R. Molecular mechanisms of drug resistance in ovarian cancer. J. Cell Physiol. 2018, 233, 4546–4562.
- Wang, J.; Seebacher, N.; Shi, H.; Kan, Q.; Duan, Z. Novel strategies to prevent the development of multidrug resistance (MDR) in cancer. Oncotarget 2017, 8, 84559–84571.
- David, W.; Chan, M.X.L.; Ngan, H.Y.S. Mechanisms of Chemoresistance in Human Ovarian Cancer at a Glance. Gynecol. Obstet. 2012, 2, e104.
- Gao, B.; Yang, F.; Chen, W.; Li, R.; Hu, X.; Liang, Y.; Li, D. Multidrug resistance affects the prognosis of primary epithelial ovarian cancer. Oncol. Lett. 2019, 18, 4262–4269.
- Cole, S.P.C. Targeting Multidrug Resistance Protein 1 (MRP1, ABCC1 ): Past, Present, and Future. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 95–117.
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464.
- Li, Q.; Shu, Y. Role of solute carriers in response to anticancer drugs. Mol. Cell. Ther. 2014, 2, 15.
- Januchowski, R.; Sterzyńska, K.; Zaorska, K.; Sosińska, P.; Klejewski, A.; Brązert, M.; Nowicki, M.; Zabel, M. Analysis of MDR genes expression and cross-resistance in eight drug resistant ovarian cancer cell lines. J. Ovarian Res. 2016, 9, 65.
- Lancaster, C.S.; Sprowl, J.A.; Walker, A.L.; Hu, S.; Gibson, A.A.; Sparreboom, A. Modulation of OATP1B-type transporter function alters cellular uptake and disposition of platinum chemotherapeutics. Mol. Cancer Ther. 2013, 12, 1537–1544.
- Siu, M.K.Y.; Kong, D.S.H.; Chan, H.Y.; Wong, E.S.Y.; Ip, P.P.-C.; Jiang, L.; Ngan, H.Y.S.; Le, X.-F.; Cheung, A.N.Y. Paradoxical Impact of Two Folate Receptors, FRα and RFC, in Ovarian Cancer: Effect on Cell Proliferation, Invasion and Clinical Outcome. PLoS ONE 2012, 7, e47201.
- Vergote, I.B.; Marth, C.; Coleman, R.L. Role of the folate receptor in ovarian cancer treatment: Evidence, mechanism, and clinical implications. Cancer Metastasis Rev. 2015, 34, 41–52.
- Wallace-Povirk, A.; Hou, Z.; Nayeen, J.; Gangjee, A.; Matherly, L.H. Folate Transport and One-Carbon Metabolism in Targeted Therapies of Epithelial Ovarian Cancer. Cancers 2021, 14, 191.
- Feng, Q.; Li, X.; Sun, W.; Sun, M.; Li, Z.; Sheng, H.; Xie, F.; Zhang, S.; Shan, C. Targeting G6PD reverses paclitaxel resistance in ovarian cancer by suppressing GSTP1. Biochem. Pharmacol. 2020, 178, 114092.
- Hollman, A.L.; Tchounwou, P.B.; Huang, H.-C. The Association between Gene-Environment Interactions and Diseases Involving the Human GST Superfamily with SNP Variants. Int. J. Environ. Res. Public Health 2016, 13, 379.
- Board, P.G.; Menon, D. Glutathione transferases, regulators of cellular metabolism and physiology. Biochim. Biophys. Acta (BBA)—Gen. Subj. 2013, 1830, 3267–3288.
- Pljesa-Ercegovac, M.; Savic-Radojevic, A.; Matic, M.; Coric, V.; Djukic, T.; Radic, T.; Simic, T. Glutathione Transferases: Potential Targets to Overcome Chemoresistance in Solid Tumors. Int. J. Mol. Sci. 2018, 19, 3785.
- McIlwain, C.C.; Townsend, D.M.; Tew, K.D. Glutathione S-transferase polymorphisms: Cancer incidence and therapy. Oncogene 2006, 25, 1639–1648.
- Oakley, A. Glutathione transferases: A structural perspective. Drug Metab. Rev. 2011, 43, 138–151.
- Board, P.; Coggan, M.; Johnston, P.; Ross, V.; Suzuki, T.; Webb, G. Genetic heterogeneity of the human glutathione transferases: A complex of gene families. Pharmacol. Ther. 1990, 48, 357–369.
- Ambrosone, C.B.; Sweeney, C.; Coles, B.F.; Thompson, A.P.; McClure, G.Y.; Korourian, S.; Fares, M.Y.; Stone, A.; Kadlubar, F.F.; Hutchins, L.F. Polymorphisms in glutathione S-transferases (GSTM1 and GSTT1) and survival after treatment for breast cancer. Cancer Res. 2001, 61, 7130–7135.
- Lo, H.-W.; Ali-Osman, F. Genetic polymorphism and function of glutathione S-transferases in tumor drug resistance. Curr. Opin. Pharmacol. 2007, 7, 367–374.
- Laborde, E. Glutathione transferases as mediators of signaling pathways involved in cell proliferation and cell death. Cell Death Differ. 2010, 17, 1373–1380.
- Tew, K.D.; Townsend, D.M. Glutathione-s-transferases as determinants of cell survival and death. Antioxid. Redox Signal. 2012, 17, 1728–1737.
- Coric, V.M.; Simic, T.P.; Pekmezovic, T.D.; Basta-Jovanovic, G.M.; Savic-Radojevic, A.R.; Radojevic-Skodric, S.M.; Matic, M.G.; Suvakov, S.R.; Dragicevic, D.P.; Radic, T.M.; et al. GSTM1 genotype is an independent prognostic factor in clear cell renal cell carcinoma. Urol. Oncol. Semin. Orig. Investig. 2017, 35, 409–417.
- Bartolini, D.; Galli, F. The functional interactome of GSTP: A regulatory biomolecular network at the interface with the Nrf2 adaption response to oxidative stress. J. Chromatogr. B 2016, 1019, 29–44.
- O’Brien, M.; Kruh, G.D.; Tew, K.D. The influence of coordinate overexpression of glutathione phase II detoxification gene products on drug resistance. J. Pharmacol. Exp. Ther. 2000, 294, 480–487.
- Smitherman, P.K.; Townsend, A.J.; Kute, T.E.; Morrow, C.S. Role of multidrug resistance protein 2 (MRP2, ABCC2) in alkylating agent detoxification: MRP2 potentiates glutathione S-transferase A1-1-mediated resistance to chlorambucil cytotoxicity. J. Pharmacol. Exp. Ther. 2004, 308, 260–267.
- Depeille, P.; Cuq, P.; Mary, S.; Passagne, I.; Evrard, A.; Cupissol, D.; Vian, L. Glutathione S-transferase M1 and multidrug resistance protein 1 act in synergy to protect melanoma cells from vincristine effects. Mol. Pharmacol. 2004, 65, 897–905.
More
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
2 times
(View History)
Update Date:
28 Nov 2022
Table of Contents




Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No