 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Muhammad Ahmad | -- | 2486 | 2022-11-24 09:40:17 | | | |
| 2 | Jessie Wu | Meta information modification | 2486 | 2022-11-25 03:16:52 | | | | |
| 3 | Jessie Wu | + 20 word(s) | 2506 | 2022-11-25 03:58:42 | | | | |
| 4 | Jessie Wu | Meta information modification | 2506 | 2022-11-25 04:19:35 | | | | |
| 5 | Muhammad Ahmad | -28 word(s) | 2478 | 2025-07-17 08:44:03 | | |
Video Upload Options
Prion-like proteins and prions (PrPC), the prion protein cellular form, shares 90% of its amino acid sequence with other mammalian proteins. PrPCs are expressed almost in all tissues of an organism, but a higher amount of PrPCs has been found in the central nervous system (CNS), particularly the synaptic membranes and PrPC are linked. Scrapie PrP (PrPSc), a mutant cellular prion protein with an altered structure, is assumed to be the key etiological cause of prion diseases. Cancers are worldwide health concerns, whether they are sporadic or hereditary. The fundamental mechanism that causes somatic or oncogenic mutations and ultimately aids cancer development is still unknown. However, mammalian cells with protein-only somatic inheritance may also contribute to cancerous malignancies. Emerging data from a recent study show that prion-like proteins and prions (PrPC) are crucial entities that have a functional role in developing neurological disorders and cancer. Furthermore, excessive PrPC expression profiling has also been detected in non-neuronal tissues, such as the lymphoid cells, kidney, GIT, lung, muscle, and mammary glands. PrPC expression is strongly linked with the proliferation and metastasis of pancreatic, prostate, colorectal, and breast malignancies. Experimental investigation presented that the PrPC expression, including the prion protein-coding gene (PRNP) and p53 ag are directly associated with tumorigenicity and metastasis (tumor suppressor gene). The ERK2 (extracellular signal-regulated kinase) pathway also confers a robust metastatic capability for PrPC-induced epithelial to mesenchymal transition. Additionally, prions could alter the epigenetic regulation of genes and overactive the mitogen-activated protein kinase (MAPK) signaling pathway, which promotes the development of cancer in humans.
1. Introduction
2. Boosts Proliferation of Cancer Cells
3. Prion-Like Proteins and Prions Encourages Cancer Cells to Invade and Spread
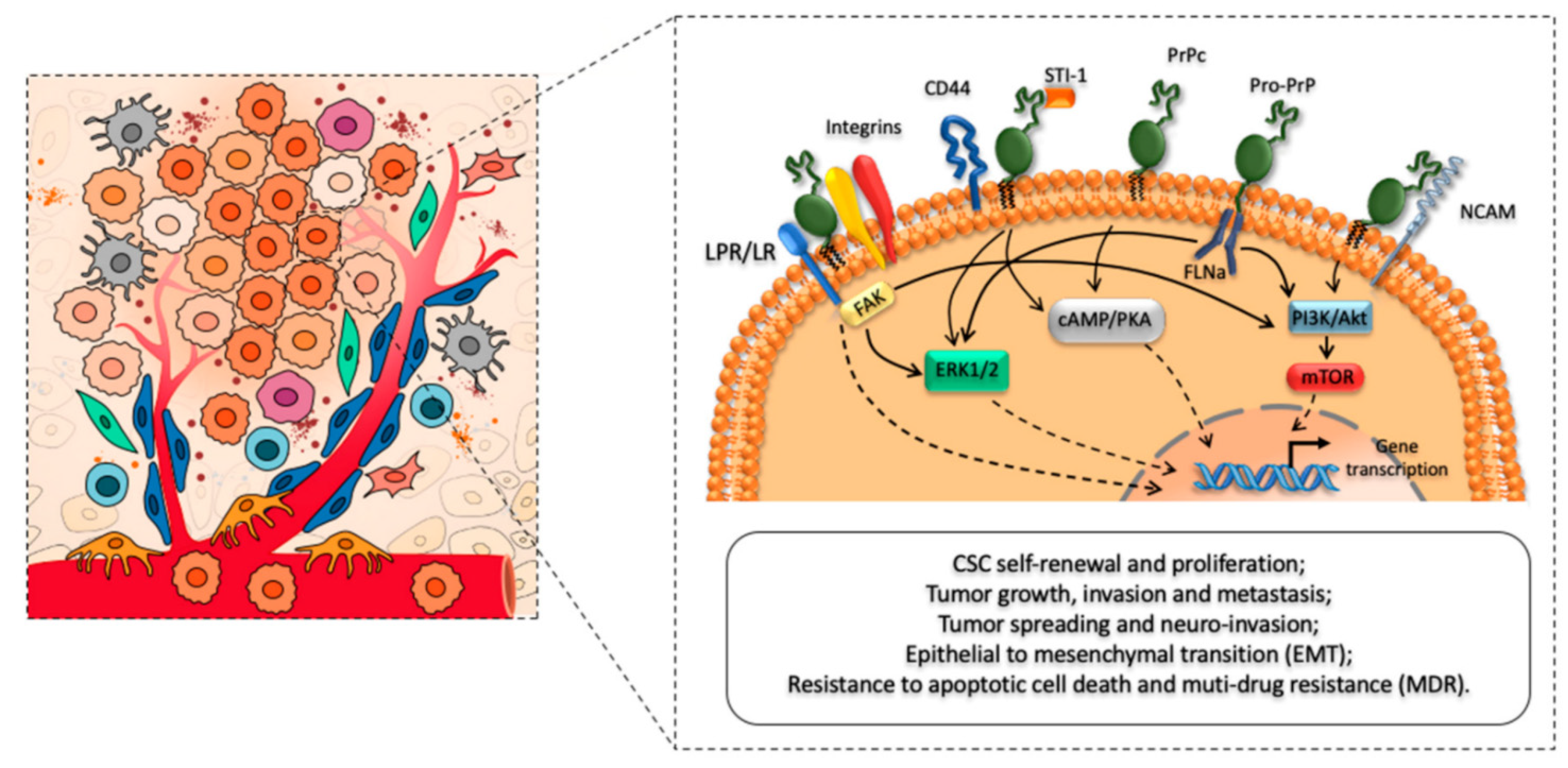
4. Prion-Like Proteins and Prions Fosters Drug Resistance in Cancer Cells
5. Prion-Like Proteins and Prions as a Potential Cancer Biomarker
6. Targeting Prion-Like Proteins and Prions as a Cancer Treatment
References
- Gao, Z.; Peng, M.; Chen, L.; Yang, X.; Li, H.; Shi, R.; Wu, G.; Cai, L.; Song, Q.; Li, C. Prion Protein Protects Cancer Cells against Endoplasmic Reticulum Stress Induced Apoptosis. Virol. Sin. 2019, 34, 222–234.
- Linden, R. The biological function of the prion protein: A cell surface scaffold of signaling modules. Front. Mol. Neurosci. 2017, 10, 77.
- Mouillet-Richard, S.; Ghazi, A.; Laurent-Puig, P. The cellular prion protein and the hallmarks of cancer. Cancers. 2021, 13, 5032.
- Hinton, C.; Antony, H.; Hashimi, S.; Munn, A.; Wei, M. Significance of Prion and Prion-Like Proteins in Cancer Development, Progression and Multi-Drug Resistance. Curr. Cancer Drug Targets 2013, 13, 895–904.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Krammer, C.; Suhre, M.H.; Kremmer, E.; Diemer, C.; Hess, S.; Schätzl, H.M.; Scheibel, T.; Vorberg, I. Prion protein/protein interactions: Fusion with yeast Sup35p—NM modulates cytosolic PrP aggregation in mammalian cells. FASEB J. 2008, 22, 762–773.
- Antony, H.; Wiegmans, A.P.; Wei, M.Q.; Chernoff, Y.O.; Khanna, K.K.; Munn, A.L. Potential roles for prions and protein-only inheritance in cancer. Cancer Metastasis Rev. 2012, 31, 1–19.
- Krance, S.H.; Luke, R.; Shenouda, M.; Israwi, A.R.; Colpitts, S.J.; Darwish, L.; Strauss, M.; Watts, J.C. Cellular models for discovering prion disease therapeutics: Progress and challenges. J. Neurochem. 2020, 153, 150–172.
- Lee, J.H.; Yun, C.W.; Han, Y.S.; Kim, S.M.; Jeong, D.; Kwon, H.Y.; Kim, H.; Baek, M.J.; Lee, S.H. Melatonin and 5-fluorouracil co-suppress colon cancer stem cells by regulating cellular prion protein-Oct4 axis. J. Pineal Res. 2018, 65, e12519.
- Mabbott, N.A. Immunology of Prion Protein and Prions. Prog. Mol. Biol. Transl. Sci. 2017, 150, 203–240.
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890.
- Llorens, F.; Ferrer, I.; Del Río, J.A. Gene expression resulting from PrPC ablation and PrPC overexpression in murine and cellular models. Mol. Neurobiol. 2014, 49, 413–423.
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144.
- Zahreddine, H.; Borden, K.L.B. Mechanisms and insights into drug resistance in cancer. Front. Pharmacol. 2013, 4 MAR, 1–8.
- Morel, E.; Fouquet, S.; Strup-Perrot, C.; Thievend, C.P.; Petit, C.; Loew, D.; Faussat, A.M.; Yvernault, L.; Pinçon-Raymond, M.; Chambaz, J.; et al. The cellular prion protein PrPc is involved in the proliferation of epithelial cells and in the distribution of junction-associated proteins. PLoS ONE 2008, 3, e3000.
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The different mechanisms of cancer drug resistance: A brief review. Adv. Pharm. Bull. 2017, 7, 339–348.
- Gasperini, L.; Legname, G. Prion protein and aging. Front. Cell Dev. Biol. 2014, 2, 44.
- Liang, J.; Pan, Y.; Zhang, D.; Guo, C.; Shi, Y.; Wang, J.; Chen, Y.; Wang, X.; Liu, J.; Guo, X.; et al. Cellular prion protein promotes proliferation and G1/S transition of human gastric cancer cells SGC7901 and AGS. FASEB J. 2007, 21, 2247–2256.
- Ryskalin, L.; Biagioni, F.; Busceti, C.L.; Giambelluca, M.A.; Morelli, L.; Frati, A.; Fornai, F. The role of cellular prion protein in promoting stemness and differentiation in cancer. Cancers 2021, 13, 170.
- Aguzzi, A. Prions and the Immune System: A Journey Through Gut, Spleen, and Nerves. Adv. Immunol. 2003, 81, 123–171.
- Málaga-Trillo, E.; Sempou, E. PrPs: Proteins with a purpose-Lessons from the zebrafish. Prion 2009, 3, 129–133.
- Colby, D.W.; Prusiner, S.B. Prions. Cold Spring Harb. Perspect. Biol. 2011, 3, 1–22.
- Lee, J.H.; Yun, C.W.; Lee, S.H. Cellular prion protein enhances drug resistance of colorectal cancer cells via regulation of a survival signal pathway. Biomol. Ther. 2018, 26, 313–321.
- Wei, W.; Shi, Q.; Zhang, N.S.; Xiao, K.; Chen, L.N.; Yang, X.D.; Ji, J.F.; Dong, X.P. Expression of prion protein is closely associated with pathological and clinical progression and abnormalities of p53 in head and neck squamous cell carcinomas. Oncol. Rep. 2016, 35, 817–824.
- Yamakawa-Kakuta, Y.; Kawamata, H.; Fujimori, T.; Imai, Y. Does the expression of HPV16/18 E6/E7 in head and neck squamous cell carcinomas relate to their clinicopathological characteristics? Int. J. Oncol. 2009, 35, 983–988.
- Wickner, R.B.; Edskes, H.K.; Shewmaker, F.; Nakayashiki, T. Prions of fungi: Inherited structures and biological roles. Nat. Rev. Microbiol. 2007, 5, 611–618.
- Liang, J.; Ge, F.; Guo, C.; Luo, G.; Wang, X.; Han, G.; Zhang, D.; Wang, J.; Li, K.; Pan, Y.; et al. Inhibition of PI3K/Akt partially leads to the inhibition of PrP C-induced drug resistance in gastric cancer cells. FEBS J. 2009, 276, 685–694.
- Baskar, R.; Dai, J.; Wenlong, N.; Yeo, R.; Yeoh, K.W. Biological response of cancer cells to radiation treatment. Front. Mol. Biosci. 2014, 1, 1–9.
- Yang, L.; Gao, Z.; Hu, L.; Wu, G.; Yang, X.; Zhang, L.; Zhu, Y.; Wong, B.S.; Xin, W.; Sy, M.S.; et al. Erratum: Glycosylphosphatidylinositol anchor modification machinery deficiency is responsible for the formation of pro-prion protein (PrP) in BxPC-3 cells and increases cancer cell motility. J. Biol. Chem. 2016, 291, 6785.
- Minikel, E.V.; Karczewski, K.J.; Martin, H.C.; Cummings, B.B.; Whiffin, N.; Rhodes, D.; Alföldi, J.; Trembath, R.C.; van Heel, D.A.; Daly, M.J.; et al. Evaluating drug targets through human loss-of-function genetic variation. Nature 2020, 581, 459–464.
- Li, C.; Yu, S.; Nakamura, F.; Yin, S.; Xu, J.; Petrolla, A.A.; Singh, N.; Tartakoff, A.; Abbott, D.W.; Xin, W.; et al. Binding of pro-prion to filamin A disrupts cytoskeleton and correlates with poor prognosis in pancreatic cancer. J. Clin. Invest. 2009, 119, 2725–2736.
- Vassallo, N.; Herms, J.; Behrens, C.; Krebs, B.; Saeki, K.; Onodera, T.; Windl, O.; Kretzschmar, H.A. Activation of phosphatidylinositol 3-kinase by cellular prion protein and its role in cell survival. Biochem. Biophys. Res. Commun. 2005, 332, 75–82.
- Rountree, R.B.; Mandl, S.J.; Nachtwey, J.M.; Dalpozzo, K.; Do, L.; Lombardo, J.R.; Schoonmaker, P.L.; Brinkmann, K.; Dirmeier, U.; Laus, R.; et al. Exosome targeting of tumor antigens expressed by cancer vaccines can improve antigen immunogenicity and therapeutic efficacy. Cancer Res. 2011, 71, 5235–5244.
- Weise, J.; Sandau, R.; Schwarting, S.; Crome, O.; Wrede, A.; Schulz-Schaeffer, W.; Zerr, I.; Bähr, M. Deletion of cellular prion protein results in reduced Akt activation, enhanced postischemic caspase-3 activation, and exacerbation of ischemic brain injury. Stroke 2006, 37, 1296–1300.
- Go, G.; Yun, C.W.; Yoon, Y.M.; Lim, J.H.; Lee, J.H.; Lee, S.H. Role of PrPC in cancer stem cell characteristics and drug resistance in colon cancer cells. Anticancer Res. 2020, 40, 5611–5620.
- Castle, A.R.; Gill, A.C. Physiological functions of the cellular prion protein. Front. Mol. Biosci. 2017, 4, 19.
- Bisht, S.; Nigam, M.; Kunjwal, S.S.; Sergey, P.; Mishra, A.P.; Sharifi-Rad, J. Cancer Stem Cells: From an Insight into the Basics to Recent Advances and Therapeutic Targeting. Stem Cells Int. 2022, 2022, 9653244.
- Cheng, Y.; Tao, L.; Xu, J.; Li, Q.; Yu, J.; Jin, Y.; Chen, Q.; Xu, Z.; Zou, Q.; Liu, X. CD44/Cellular prion protein interact in multidrug resistant breast cancer cells and correlate with responses to neoadjuvant chemotherapy in breast cancer patients. Mol. Carcinog. 2014, 53, 686–697.
- Antonacopoulou, A.G.; Grivas, P.D.; Skarlas, L.; Kalofonos, M.; Scopa, C.D.; Kalofonos, H.P. POLR2F, ATP6V0A1 and PRNP expression in colorectal cancer: New molecules with prognostic significance? Anticancer Res. 2008, 28, 1221–1227.
- Lu, W.; Kang, Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev. Cell 2019, 49, 361–374.
- Yu, G.; Jiang, L.; Xu, Y.; Guo, H.; Liu, H.; Zhang, Y.; Yang, H.; Yuan, C.; Ma, J. Silencing Prion Protein in MDA-MB-435 Breast Cancer Cells Leads to Pleiotropic Cellular Responses to Cytotoxic Stimuli. PLoS ONE 2012, 7, e41771.
- Krammer, C.; Kryndushkin, D.; Suhre, M.H.; Kremmer, E.; Hofmann, A.; Pfeifer, A.; Scheibel, T.; Wickner, R.B.; Schätzl, H.M.; Vorberg, I. The yeast Sup35NM domain propagates as a prion in mammalian cells. Proc. Natl. Acad. Sci. USA 2009, 106, 462–467.
- Meslin, F.; Conforti, R.; Mazouni, C.; Morel, N.; Tomasic, G.; Drusch, F.; Yacoub, M.; Sabourin, J.C.; Grassi, J.; Delaloge, S.; et al. Efficacy of adjuvant chemotherapy according to Prion protein expression in patients with estrogen receptor-negative breast cancer. Ann. Oncol. 2007, 18, 1793–1798.
- Park, J.Y.; Jeong, J.K.; Lee, J.H.; Moon, J.H.; Kim, S.W.; Lee, Y.J.; Park, S.Y. Induction of cellular prion protein (PrPc) under hypoxia inhibits apoptosis caused by TRAIL treatment. Oncotarget 2015, 6, 5342–5353.
- Ding, M.; Chen, Y.; Lang, Y.; Cui, L. The Role of Cellular Prion Protein in Cancer Biology: A Potential Therapeutic Target. Front. Oncol. 2021, 11, 3610.
- Wang, Y.J.; Herlyn, M. The emerging roles of Oct4 in tumor-initiating cells. Am. J. Physiol.—Cell Physiol. 2015, 309, C709–C718.

