 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Vivi Li | -- | 1605 | 2022-11-24 01:46:32 |
Video Upload Options
In medicine, proteopathy (/proʊtiːˈɒpəθiː/; from proteo- [pref. protein]; -pathy [suff. disease]; proteopathies pl.; proteopathic adj) refers to a class of diseases in which certain proteins become structurally abnormal, and thereby disrupt the function of cells, tissues and organs of the body. Often the proteins fail to fold into their normal configuration; in this misfolded state, the proteins can become toxic in some way (a toxic gain-of-function) or they can lose their normal function. The proteopathies (also known as proteinopathies, protein conformational disorders, or protein misfolding diseases) include such diseases as Creutzfeldt–Jakob disease and other prion diseases, Alzheimer's disease, Parkinson's disease, amyloidosis, multiple system atrophy, and a wide range of other disorders. The term proteopathy was first proposed in 2000 by Lary Walker and Harry LeVine. The concept of proteopathy can trace its origins to the mid-19th century, when, in 1854, Rudolf Virchow coined the term amyloid ("starch-like") to describe a substance in cerebral corpora amylacea that exhibited a chemical reaction resembling that of cellulose. In 1859, Friedreich and Kekulé demonstrated that, rather than consisting of cellulose, "amyloid" actually is rich in protein. Subsequent research has shown that many different proteins can form amyloid, and that all amyloids show birefringence in cross-polarized light after staining with the dye Congo red, as well as a fibrillar ultrastructure when viewed with an electron microscope. However, some proteinaceous lesions lack birefringence and contain few or no classical amyloid fibrils, such as the diffuse deposits of amyloid beta (Aβ) protein in the brains of people with Alzheimer's. Furthermore, evidence has emerged that small, non-fibrillar protein aggregates known as oligomers are toxic to the cells of an affected organ, and that amyloidogenic proteins in their fibrillar form may be relatively benign.
1. Pathophysiology
In most, if not all proteopathies, a change in 3-dimensional folding (conformation) increases the tendency of a specific protein to bind to itself.[1] In this aggregated form, the protein is resistant to clearance and can interfere with the normal capacity of the affected organs. In some cases, misfolding of the protein results in a loss of its usual function. For example, cystic fibrosis is caused by a defective cystic fibrosis transmembrane conductance regulator (CFTR) protein,[2] and in amyotrophic lateral sclerosis / frontotemporal lobar degeneration (FTLD), certain gene-regulating proteins inappropriately aggregate in the cytoplasm, and thus are unable to perform their normal tasks within the nucleus.[3][4] Because proteins share a common structural feature known as the polypeptide backbone, all proteins have the potential to misfold under some circumstances.[5] However, only a relatively small number of proteins are linked to proteopathic disorders, possibly due to structural idiosyncrasies of the vulnerable proteins. For example, proteins that are normally unfolded or relatively unstable as monomers (that is, as single, unbound protein molecules) are more likely to misfold into an abnormal conformation.[1][5][6] In nearly all instances, the disease-causing molecular configuration involves an increase in beta-sheet secondary structure of the protein.[1][5][7][8][9] The abnormal proteins in some proteopathies have been shown to fold into multiple 3-dimensional shapes; these variant, proteinaceous structures are defined by their different pathogenic, biochemical, and conformational properties.[10] They have been most thoroughly studied with regard to prion disease, and are referred to as protein strains.[11][12]
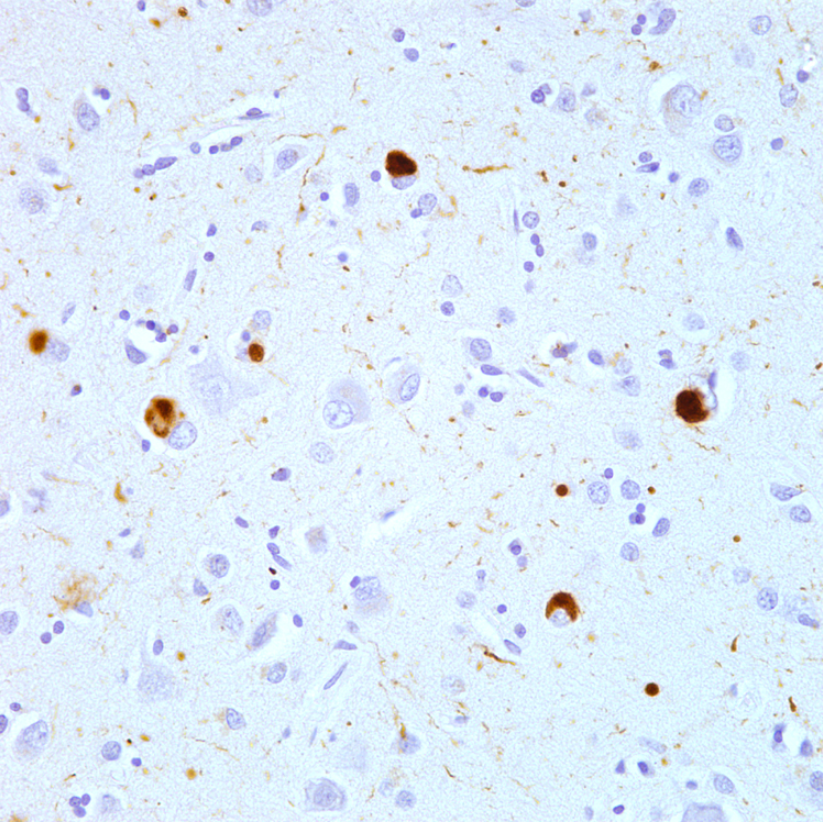
The likelihood that proteopathy will develop is increased by certain risk factors that promote the self-assembly of a protein. These include destabilizing changes in the primary amino acid sequence of the protein, post-translational modifications (such as hyperphosphorylation), changes in temperature or pH, an increase in production of a protein, or a decrease in its clearance.[1][5][13] Advancing age is a strong risk factor,[13] as is traumatic brain injury.[14][15] In the aging brain, multiple proteopathies can overlap.[16] For example, in addition to tauopathy and Aβ-amyloidosis (which coexist as key pathologic features of Alzheimer's disease), many Alzheimer patients have concomitant synucleinopathy (Lewy bodies) in the brain.[17]
It is hypothesized that chaperones and co-chaperones (proteins that assist protein folding) may antagonize proteotoxicity during aging and in protein misfolding-diseases to maintain proteostasis.[18][19][20]
2. Seeded Induction
Some proteins can be induced to form abnormal assemblies by exposure to the same (or similar) protein that has folded into a disease-causing conformation, a process called 'seeding' or 'permissive templating'.[21][22] In this way, the disease state can be brought about in a susceptible host by the introduction of diseased tissue extract from an afflicted donor. The best known forms of inducible proteopathy are prion diseases,[23] which can be transmitted by exposure of a host organism to purified prion protein in a disease-causing conformation.[24][25] There is now evidence that other proteopathies can be induced by a similar mechanism, including Aβ amyloidosis, amyloid A (AA) amyloidosis, and apolipoprotein AII amyloidosis,[22][26] tauopathy,[27] synucleinopathy,[28][29][30][31] and the aggregation of superoxide dismutase-1 (SOD1),[32][33] polyglutamine,[34][35] and TAR DNA-binding protein-43 (TDP-43).[36]
In all of these instances, an aberrant form of the protein itself appears to be the pathogenic agent. In some cases, the deposition of one type of protein can be experimentally induced by aggregated assemblies of other proteins that are rich in β-sheet structure, possibly because of structural complementarity of the protein molecules. For example, AA amyloidosis can be stimulated in mice by such diverse macromolecules as silk, the yeast amyloid Sup35, and curli fibrils from the bacterium Escherichia coli.[37] AII amyloid can be induced in mice by a variety of β-sheet rich amyloid fibrils,[38] and cerebral tauopathy can be induced by brain extracts that are rich in aggregated Aβ.[39] There is also experimental evidence for cross-seeding between prion protein and Aβ.[40] In general, such heterologous seeding is less efficient than is seeding by a corrupted form of the same protein.
3. List of Proteopathies
| Proteopathy | Major aggregating protein |
| Alzheimer's disease[6] | Amyloid β peptide (Aβ); Tau protein (see tauopathies) |
| Cerebral β-amyloid angiopathy[41] | Amyloid β peptide (Aβ) |
| Retinal ganglion cell degeneration in glaucoma[42] | Amyloid β peptide (Aβ) |
| Prion diseases (multiple)[43] | Prion protein |
| Parkinson's disease and other synucleinopathies (multiple)[44] | α-Synuclein |
| Tauopathies (multiple)[45] | Microtubule-associated protein tau (Tau protein) |
| Frontotemporal lobar degeneration (FTLD) (Ubi+, Tau-)[46] | TDP-43 |
| FTLD–FUS[46] | Fused in sarcoma (FUS) protein |
| Amyotrophic lateral sclerosis (ALS)[47][48] | Superoxide dismutase, TDP-43, FUS, C9ORF72, ubiquilin-2 (UBQLN2) |
| Huntington's disease and other trinucleotide repeat disorders (multiple)[49][50] | Proteins with tandem glutamine expansions |
| Familial British dementia[41] | ABri |
| Familial Danish dementia[41] | ADan |
| Hereditary cerebral hemorrhage with amyloidosis (Icelandic) (HCHWA-I)[41] | Cystatin C |
| CADASIL[51] | Notch3 |
| Alexander disease[52] | Glial fibrillary acidic protein (GFAP) |
| Pelizaeus-Merzbacher disease | proteolipid protein (PLP) |
| Seipinopathies[53] | Seipin |
| Familial amyloidotic neuropathy, Senile systemic amyloidosis | Transthyretin[54] |
| Serpinopathies (multiple)[55] | Serpins |
| AL (light chain) amyloidosis (primary systemic amyloidosis) | Monoclonal immunoglobulin light chains[54] |
| AH (heavy chain) amyloidosis | Immunoglobulin heavy chains[54] |
| AA (secondary) amyloidosis | Amyloid A protein[54] |
| Type II diabetes[56] | Islet amyloid polypeptide (IAPP; amylin) |
| Aortic medial amyloidosis | Medin (lactadherin)[54] |
| ApoAI amyloidosis | Apolipoprotein AI[54] |
| ApoAII amyloidosis | Apolipoprotein AII[54] |
| ApoAIV amyloidosis | Apolipoprotein AIV[54] |
| Familial amyloidosis of the Finnish type (FAF) | Gelsolin[54] |
| Lysozyme amyloidosis | Lysozyme[54] |
| Fibrinogen amyloidosis | Fibrinogen[54] |
| Dialysis amyloidosis | Beta-2 microglobulin[54] |
| Inclusion body myositis/myopathy[57] | Amyloid β peptide (Aβ) |
| Cataracts[58] | Crystallins |
| Retinitis pigmentosa with rhodopsin mutations[59] | rhodopsin |
| Medullary thyroid carcinoma | Calcitonin[54] |
| Cardiac atrial amyloidosis | Atrial natriuretic factor[54] |
| Pituitary prolactinoma | Prolactin[54] |
| Hereditary lattice corneal dystrophy | Keratoepithelin[54] |
| Cutaneous lichen amyloidosis[60] | Keratins |
| Mallory bodies[61] | Keratin intermediate filament proteins |
| Corneal lactoferrin amyloidosis | Lactoferrin[54] |
| Pulmonary alveolar proteinosis | Surfactant protein C (SP-C)[54] |
| Odontogenic (Pindborg) tumor amyloid | Odontogenic ameloblast-associated protein[54] |
| Seminal vesicle amyloid | Semenogelin I[54] |
| Apolipoprotein C2 amyloidosis | Apolipoprotein C2 (ApoC2)[54] |
| Apolipoprotein C3 amyloidosis | Apolipoprotein C3 (ApoC3)[54] |
| Lect2 amyloidosis | Leukocyte chemotactic factor-2 (Lect2)[54] |
| Insulin amyloidosis | Insulin[54] |
| Galectin-7 amyloidosis (primary localized cutaneous amyloidosis) | Galectin-7 (Gal7)[54] |
| Corneodesmosin amyloidosis | Corneodesmosin[54] |
| Enfuvirtide amyloidosis[62] | Enfuvirtide[54] |
| Cystic fibrosis[63] | cystic fibrosis transmembrane conductance regulator (CFTR) protein |
| Sickle cell disease[64] | Hemoglobin |
4. Management
The development of effective treatments for many proteopathies has been challenging.[65][66] Because the proteopathies often involve different proteins arising from different sources, treatment strategies must be customized to each disorder; however, general therapeutic approaches include maintaining the function of affected organs, reducing the formation of the disease-causing proteins, preventing the proteins from misfolding and/or aggregating, or promoting their removal.[65][67][68] For example, in Alzheimer's disease, researchers are seeking ways to reduce the production of the disease-associated protein Aβ by inhibiting the enzymes that free it from its parent protein.[66] Another strategy is to use antibodies to neutralize specific proteins by active or passive immunization.[69] In some proteopathies, inhibiting the toxic effects of protein oligomers might be beneficial.[70] Amyloid A (AA) amyloidosis can be reduced by treating the inflammatory state that increases the amount of the protein in the blood (referred to as serum amyloid A, or SAA).[65] In immunoglobulin light chain amyloidosis (AL amyloidosis), chemotherapy can be used to lower the number of the blood cells that make the light chain protein that forms amyloid in various bodily organs.[71] Transthyretin (TTR) amyloidosis (ATTR) results from the deposition of misfolded TTR in multiple organs.[72] Because TTR is mainly produced in the liver, TTR amyloidosis can be slowed in some hereditary cases by liver transplantation.[73] TTR amyloidosis also can be treated by stabilizing the normal assemblies of the protein (called tetramers because they consist of four TTR molecules bound together). Stabilization prevents individual TTR molecules from escaping, misfolding, and aggregating into amyloid.[74][75]
Several other treatment strategies for proteopathies are being investigated, including small molecules and biologic medicines such as small interfering RNAs, antisense oligonucleotides, peptides, and engineered immune cells.[71][74][76][77] In some cases, multiple therapeutic agents may be combined to improve effectiveness.[71][78]
5. Additional Images
-
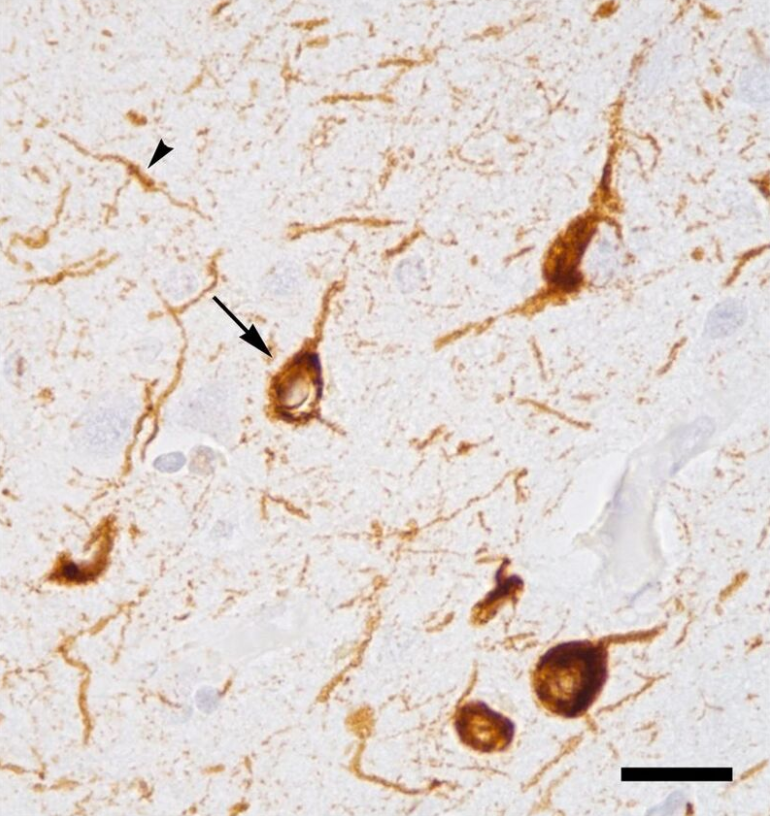
Micrograph of tauopathy (brown) in a neuronal cell body (arrow) and process (arrowhead) in the cerebral cortex of a patient with Alzheimer's disease. Bar = 25 microns (0.025mm). https://handwiki.org/wiki/index.php?curid=1988295
References
- "Conformational disease". Lancet 350 (9071): 134–8. July 1997. doi:10.1016/S0140-6736(97)02073-4. PMID 9228977. https://dx.doi.org/10.1016%2FS0140-6736%2897%2902073-4
- "Protein misfolding and disease: from the test tube to the organism". Current Opinion in Chemical Biology 12 (1): 25–31. February 2008. doi:10.1016/j.cbpa.2008.02.011. PMID 18295611. https://dx.doi.org/10.1016%2Fj.cbpa.2008.02.011
- "Conjoint pathologic cascades mediated by ALS/FTLD-U linked RNA-binding proteins TDP-43 and FUS". Neurology 77 (17): 1636–43. October 2011. doi:10.1212/WNL.0b013e3182343365. PMID 21956718. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3198978
- RNA binding proteins and the genesis of neurodegenerative diseases. Advances in Experimental Medicine and Biology. 822. 2015. pp. 11–5. doi:10.1007/978-3-319-08927-0_3. ISBN 978-3-319-08926-3. PMID 25416971. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4694570
- "Protein misfolding, evolution and disease". Trends in Biochemical Sciences 24 (9): 329–32. September 1999. doi:10.1016/S0968-0004(99)01445-0. PMID 10470028. https://dx.doi.org/10.1016%2FS0968-0004%2899%2901445-0
- "Self-propagation of pathogenic protein aggregates in neurodegenerative diseases". Nature 501 (7465): 45–51. September 2013. doi:10.1038/nature12481. PMID 24005412. Bibcode: 2013Natur.501...45J. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3963807
- "Folding proteins in fatal ways". Nature 426 (6968): 900–4. December 2003. doi:10.1038/nature02264. PMID 14685251. Bibcode: 2003Natur.426..900S. https://dx.doi.org/10.1038%2Fnature02264
- "The amyloid state of proteins in human diseases". Cell 148 (6): 1188–203. March 2012. doi:10.1016/j.cell.2012.02.022. PMID 22424229. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3353745
- "Label-free vibrational imaging of different Aβ plaque types in Alzheimer's disease reveals sequential events in plaque development". Acta Neuropathologica Communications 8 (1): 222. December 2020. doi:10.1186/s40478-020-01091-5. PMID 33308303. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=7733282
- "Proteopathic Strains and the Heterogeneity of Neurodegenerative Diseases". Annual Review of Genetics 50: 329–346. November 2016. doi:10.1146/annurev-genet-120215-034943. PMID 27893962. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=6690197
- "A general model of prion strains and their pathogenicity". Science 318 (5852): 930–6. November 2007. doi:10.1126/science.1138718. PMID 17991853. Bibcode: 2007Sci...318..930C. https://dx.doi.org/10.1126%2Fscience.1138718
- "De novo generation of prion strains". Nature Reviews. Microbiology 9 (11): 771–7. September 2011. doi:10.1038/nrmicro2650. PMID 21947062. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3924856
- "The cerebral proteopathies". Neurobiology of Aging 21 (4): 559–61. 2000. doi:10.1016/S0197-4580(00)00160-3. PMID 10924770. https://dx.doi.org/10.1016%2FS0197-4580%2800%2900160-3
- "Traumatic brain injury--football, warfare, and long-term effects". The New England Journal of Medicine 363 (14): 1293–6. September 2010. doi:10.1056/NEJMp1007051. PMID 20879875. https://dx.doi.org/10.1056%2FNEJMp1007051
- "The neuropathology of chronic traumatic encephalopathy". Brain Pathology 25 (3): 350–64. May 2015. doi:10.1111/bpa.12248. PMID 25904048. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4526170
- "Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature". Journal of Neuropathology and Experimental Neurology 71 (5): 362–81. May 2012. doi:10.1097/NEN.0b013e31825018f7. PMID 22487856. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3560290
- "Dementia with Lewy bodies: Definition, diagnosis, and pathogenic relationship to Alzheimer's disease". Neuropsychiatric Disease and Treatment 3 (5): 619–25. 2007. PMID 19300591. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2656298
- "Molecular chaperones antagonize proteotoxicity by differentially modulating protein aggregation pathways". Prion 3 (2): 51–8. 2009. doi:10.4161/pri.3.2.8587. PMID 19421006. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2712599
- "A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease". Cell Reports 9 (3): 1135–50. November 2014. doi:10.1016/j.celrep.2014.09.042. PMID 25437566. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4255334
- "Model systems of protein-misfolding diseases reveal chaperone modifiers of proteotoxicity". Disease Models & Mechanisms 9 (8): 823–38. August 2016. doi:10.1242/dmm.024703. PMID 27491084. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=5007983
- "Expression of normal sequence pathogenic proteins for neurodegenerative disease contributes to disease risk: 'permissive templating' as a general mechanism underlying neurodegeneration". Biochemical Society Transactions 33 (Pt 4): 578–81. August 2005. doi:10.1042/BST0330578. PMID 16042548. https://dx.doi.org/10.1042%2FBST0330578
- "Inducible proteopathies". Trends in Neurosciences 29 (8): 438–43. August 2006. doi:10.1016/j.tins.2006.06.010. PMID 16806508. https://dx.doi.org/10.1016%2Fj.tins.2006.06.010
- "Shattuck lecture--neurodegenerative diseases and prions". The New England Journal of Medicine 344 (20): 1516–26. May 2001. doi:10.1056/NEJM200105173442006. PMID 11357156. https://dx.doi.org/10.1056%2FNEJM200105173442006
- "From microbes to prions the final proof of the prion hypothesis". Cell 121 (2): 155–7. April 2005. doi:10.1016/j.cell.2005.04.002. PMID 15851020. https://dx.doi.org/10.1016%2Fj.cell.2005.04.002
- "The role of cofactors in prion propagation and infectivity". PLOS Pathogens 8 (4): e1002589. 2012. doi:10.1371/journal.ppat.1002589. PMID 22511864. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3325206
- "Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host". Science 313 (5794): 1781–4. September 2006. doi:10.1126/science.1131864. PMID 16990547. Bibcode: 2006Sci...313.1781M. https://dx.doi.org/10.1126%2Fscience.1131864
- "Transmission and spreading of tauopathy in transgenic mouse brain". Nature Cell Biology 11 (7): 909–13. July 2009. doi:10.1038/ncb1901. PMID 19503072. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2726961
- "Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein". Proceedings of the National Academy of Sciences of the United States of America 106 (31): 13010–5. August 2009. doi:10.1073/pnas.0903691106. PMID 19651612. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2722313
- "α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells". The Journal of Clinical Investigation 121 (2): 715–25. February 2011. doi:10.1172/JCI43366. PMID 21245577. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3026723
- "Transfer of host-derived α synuclein to grafted dopaminergic neurons in rat". Neurobiology of Disease 43 (3): 552–7. September 2011. doi:10.1016/j.nbd.2011.05.001. PMID 21600984. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3430516
- "Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease". Nature Medicine 14 (5): 504–6. May 2008. doi:10.1038/nm1747. PMID 18391962. https://dx.doi.org/10.1038%2Fnm1747
- Feany, Mel B., ed (May 2010). "Superoxide dismutase 1 and tgSOD1 mouse spinal cord seed fibrils, suggesting a propagative cell death mechanism in amyotrophic lateral sclerosis". PLOS ONE 5 (5): e10627. doi:10.1371/journal.pone.0010627. PMID 20498711. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2869360
- "Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells". Proceedings of the National Academy of Sciences of the United States of America 108 (9): 3548–53. March 2011. doi:10.1073/pnas.1017275108. PMID 21321227. Bibcode: 2011PNAS..108.3548M. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3048161
- "Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates". Nature Cell Biology 11 (2): 219–25. February 2009. doi:10.1038/ncb1830. PMID 19151706. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2757079
- "Prion-Like Characteristics of Polyglutamine-Containing Proteins". Cold Spring Harbor Perspectives in Medicine 8 (2): a024257. February 2018. doi:10.1101/cshperspect.a024257. PMID 28096245. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=5793740
- "A seeding reaction recapitulates intracellular formation of Sarkosyl-insoluble transactivation response element (TAR) DNA-binding protein-43 inclusions". The Journal of Biological Chemistry 286 (21): 18664–72. May 2011. doi:10.1074/jbc.M111.231209. PMID 21454603. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3099683
- "Protein fibrils in nature can enhance amyloid protein A amyloidosis in mice: Cross-seeding as a disease mechanism". Proceedings of the National Academy of Sciences of the United States of America 102 (17): 6098–102. April 2005. doi:10.1073/pnas.0501814102. PMID 15829582. Bibcode: 2005PNAS..102.6098L. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1087940
- "Induction of AApoAII amyloidosis by various heterogeneous amyloid fibrils". FEBS Letters 563 (1–3): 179–84. April 2004. doi:10.1016/S0014-5793(04)00295-9. PMID 15063745. https://dx.doi.org/10.1016%2FS0014-5793%2804%2900295-9
- "Induction of tau pathology by intracerebral infusion of amyloid-beta -containing brain extract and by amyloid-beta deposition in APP x Tau transgenic mice". The American Journal of Pathology 171 (6): 2012–20. December 2007. doi:10.2353/ajpath.2007.070403. PMID 18055549. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2111123
- "Molecular cross talk between misfolded proteins in animal models of Alzheimer's and prion diseases". The Journal of Neuroscience 30 (13): 4528–35. March 2010. doi:10.1523/JNEUROSCI.5924-09.2010. PMID 20357103. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2859074
- "Cerebral amyloid angiopathies: a pathologic, biochemical, and genetic view". Journal of Neuropathology and Experimental Neurology 62 (9): 885–98. September 2003. doi:10.1093/jnen/62.9.885. PMID 14533778. https://dx.doi.org/10.1093%2Fjnen%2F62.9.885
- "Targeting amyloid-beta in glaucoma treatment". Proceedings of the National Academy of Sciences of the United States of America 104 (33): 13444–9. August 2007. doi:10.1073/pnas.0703707104. PMID 17684098. Bibcode: 2007PNAS..10413444G. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1940230
- Prusiner, SB (2004). Prion Biology and Diseases (2 ed.). Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. ISBN 0-87969-693-1.
- "100 years of Lewy pathology". Nature Reviews. Neurology 9 (1): 13–24. January 2013. doi:10.1038/nrneurol.2012.242. PMID 23183883. https://dx.doi.org/10.1038%2Fnrneurol.2012.242
- "Invited review: Prion-like transmission and spreading of tau pathology". Neuropathology and Applied Neurobiology 41 (1): 47–58. February 2015. doi:10.1111/nan.12197. PMID 25399729. https://dx.doi.org/10.1111%2Fnan.12197
- "Frontotemporal lobar degeneration: Pathogenesis, pathology and pathways to phenotype". Brain Pathology 27 (6): 723–736. November 2017. doi:10.1111/bpa.12486. PMID 28100023. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=8029341
- "From molecule to molecule and cell to cell: prion-like mechanisms in amyotrophic lateral sclerosis". Neurobiology of Disease 77: 257–65. May 2015. doi:10.1016/j.nbd.2015.02.009. PMID 25701498. https://dx.doi.org/10.1016%2Fj.nbd.2015.02.009
- "Amyotrophic lateral sclerosis". Current Opinion in Neurology 25 (5): 530–5. October 2012. doi:10.1097/WCO.0b013e328356d328. PMID 22918486. https://dx.doi.org/10.1097%2FWCO.0b013e328356d328
- "Trinucleotide repeat disorders". Annual Review of Neuroscience 30 (1): 575–621. July 2007. doi:10.1146/annurev.neuro.29.051605.113042. PMID 17417937. https://dx.doi.org/10.1146%2Fannurev.neuro.29.051605.113042
- "Trinucleotide repeats: a structural perspective". Frontiers in Neurology 4: 76. 2013. doi:10.3389/fneur.2013.00076. PMID 23801983. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3687200
- "CADASIL: Notch signaling defect or protein accumulation problem?". The Journal of Clinical Investigation 105 (5): 561–2. March 2000. doi:10.1172/JCI9511. PMID 10712425. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=292459
- "GFAP and its role in Alexander disease". Experimental Cell Research 313 (10): 2077–87. June 2007. doi:10.1016/j.yexcr.2007.04.004. PMID 17498694. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2702672
- "Seipinopathy: a novel endoplasmic reticulum stress-associated disease". Brain 132 (Pt 1): 8–15. January 2009. doi:10.1093/brain/awn216. PMID 18790819. https://dx.doi.org/10.1093%2Fbrain%2Fawn216
- "Amyloid fibril proteins and amyloidosis: chemical identification and clinical classification International Society of Amyloidosis 2016 Nomenclature Guidelines". Amyloid 23 (4): 209–213. December 2016. doi:10.1080/13506129.2016.1257986. PMID 27884064. https://dx.doi.org/10.1080%2F13506129.2016.1257986
- "Serpinopathies and the conformational dementias". Nature Reviews Genetics 3 (10): 759–68. October 2002. doi:10.1038/nrg907. PMID 12360234. https://dx.doi.org/10.1038%2Fnrg907
- "Prion-Like Protein Aggregates and Type 2 Diabetes". Cold Spring Harbor Perspectives in Medicine 7 (5): a024315. May 2017. doi:10.1101/cshperspect.a024315. PMID 28159831. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=5411686
- "Inclusion-body myositis: a myodegenerative conformational disorder associated with Abeta, protein misfolding, and proteasome inhibition". Neurology 66 (2 Suppl 1): S39-48. January 2006. doi:10.1212/01.wnl.0000192128.13875.1e. PMID 16432144. https://dx.doi.org/10.1212%2F01.wnl.0000192128.13875.1e
- "Crystallin proteins and amyloid fibrils". Cellular and Molecular Life Sciences 66 (1): 62–81. January 2009. doi:10.1007/s00018-008-8327-4. PMID 18810322. https://ro.uow.edu.au/cgi/viewcontent.cgi?article=1967&context=scipapers.
- "Conformational diseases: looking into the eyes". Brain Research Bulletin 81 (1): 12–24. January 2010. doi:10.1016/j.brainresbull.2009.09.015. PMID 19808079. https://dx.doi.org/10.1016%2Fj.brainresbull.2009.09.015
- "Cytokeratins in primary cutaneous amyloidosis". The Australasian Journal of Dermatology 39 (2): 81–5. May 1998. doi:10.1111/j.1440-0960.1998.tb01253.x. PMID 9611375. https://dx.doi.org/10.1111%2Fj.1440-0960.1998.tb01253.x
- "Interaction of stress proteins with misfolded keratins". European Journal of Cell Biology 84 (2–3): 329–39. March 2005. doi:10.1016/j.ejcb.2004.12.018. PMID 15819411. https://dx.doi.org/10.1016%2Fj.ejcb.2004.12.018
- null
- "The cystic fibrosis transmembrane conductance regulator (CFTR) and its stability". Cellular and Molecular Life Sciences 74 (1): 23–38. January 2017. doi:10.1007/s00018-016-2386-8. PMID 27734094. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=5209436
- "Sickle-cell disease". Lancet 364 (9442): 1343–60. 2004. doi:10.1016/S0140-6736(04)17192-4. PMID 15474138. https://dx.doi.org/10.1016%2FS0140-6736%2804%2917192-4
- "Amyloidosis". Annu Rev Med 57: 223–241. 2006. doi:10.1146/annurev.med.57.121304.131243. PMID 16409147. https://dx.doi.org/10.1146%2Fannurev.med.57.121304.131243
- "Alzheimer's disease: the challenge of the second century". Sci Transl Med 3 (77): 77sr1. 2011. doi:10.1126/scitranslmed.3002369. PMID 21471435. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3130546
- "Pathogenesis, diagnosis and treatment of systemic amyloidosis". Phil Trans R Soc Lond B 356 (1406): 203–211. 2001. doi:10.1098/rstb.2000.0766. PMID 11260801. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1088426
- "Proteopathy: the next therapeutic frontier?". Curr Opin Investig Drugs 3 (5): 782–7. 2002. PMID 12090553. http://www.ncbi.nlm.nih.gov/pubmed/12090553
- "Vaccination strategies in tauopathies and synucleinopathies". J Neurochem 143 (5): 467–488. 2017. doi:10.1111/jnc.14207. PMID 28869766. https://dx.doi.org/10.1111%2Fjnc.14207
- "Synaptotoxic amyloid-β oligomers: a molecular basis for the cause, diagnosis, and treatment of Alzheimer's disease?". J Alzheimers Dis 33 (Suppl 1): S49-65. 2013. doi:10.3233/JAD-2012-129039. PMID 22785404. https://dx.doi.org/10.3233%2FJAD-2012-129039
- "Recent advances in understanding and treating immunoglobulin light chain amyloidosis". F1000Res 7: 1348. 2018. doi:10.12688/f1000research.15353.1. PMID 30228867. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=6117860
- "Liver transplantation in transthyretin amyloidosis: issues and challenges". Liver Transpl 21 (3): 282–292. 2015. doi:10.1002/lt.24058. PMID 25482846. https://dx.doi.org/10.1002%2Flt.24058
- "Liver transplantation for hereditary transthyretin amyloidosis". Liver Transpl 6 (3): 263–276. 2000. doi:10.1053/lv.2000.6145. PMID 10827225. https://dx.doi.org/10.1053%2Flv.2000.6145
- "Survival After Transplantation in Patients With Mutations Other Than Val30Met: Extracts From the FAP World Transplant Registry". Transplantation 100 (2): 373–381. 2016. doi:10.1097/TP.0000000000001021. PMID 26656838. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4732012
- "Mechanism of Action and Clinical Application of Tafamidis in Hereditary Transthyretin Amyloidosis". Neurol Ther 5 (1): 1–25. 2016. doi:10.1007/s40120-016-0040-x. PMID 26894299. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4919130
- "Single-stranded RNAs use RNAi to potently and allele-selectively inhibit mutant huntingtin expression". Cell 150 (5): 895–908. 2012. doi:10.1016/j.cell.2012.08.002. PMID 22939619. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3444165
- "Emerging therapeutic targets currently under investigation for the treatment of systemic amyloidosis". Expert Opin Ther Targets 21 (12): 1095–1110. 2017. doi:10.1080/14728222.2017.1398235. PMID 29076382. https://dx.doi.org/10.1080%2F14728222.2017.1398235
- "Novel Approaches for the Management of AL Amyloidosis". Curr Hematol Malig Rep 13 (3): 212–219. 2018. doi:10.1007/s11899-018-0450-1. PMID 29951831. https://dx.doi.org/10.1007%2Fs11899-018-0450-1


