Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Abhinav Ram Mohan | -- | 5167 | 2022-11-22 17:43:00 | | | |
| 2 | Peter Tang | Meta information modification | 5167 | 2022-11-24 03:21:22 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Mohan, A.R.; Wang, Q.; Dhapare, S.; Bielski, E.; Kaviratna, A.; Han, L.; Boc, S.; Newman, B. Dry Powder Inhaler Products. Encyclopedia. Available online: https://encyclopedia.pub/entry/36127 (accessed on 04 August 2026).
Mohan AR, Wang Q, Dhapare S, Bielski E, Kaviratna A, Han L, et al. Dry Powder Inhaler Products. Encyclopedia. Available at: https://encyclopedia.pub/entry/36127. Accessed August 04, 2026.
Mohan, Abhinav Ram, Qiang Wang, Sneha Dhapare, Elizabeth Bielski, Anubhav Kaviratna, Liangfeng Han, Susan Boc, Bryan Newman. "Dry Powder Inhaler Products" Encyclopedia, https://encyclopedia.pub/entry/36127 (accessed August 04, 2026).
Mohan, A.R., Wang, Q., Dhapare, S., Bielski, E., Kaviratna, A., Han, L., Boc, S., & Newman, B. (2022, November 23). Dry Powder Inhaler Products. In Encyclopedia. https://encyclopedia.pub/entry/36127
Mohan, Abhinav Ram, et al. "Dry Powder Inhaler Products." Encyclopedia. Web. 23 November, 2022.
Copy Citation
Dry powder inhalers (DPIs) are drug–device combination products where the complexity of the formulation, its interaction with the device, and input from users play important roles in the drug delivery. As the landscape of DPI products advances with new powder formulations and novel device designs, understanding how these advancements impact performance can aid in developing generics that are therapeutically equivalent to the reference listed drug (RLD) products.
dry powder inhaler (DPI)
orally inhaled drug products (OIDP)
bioequivalence (BE)
generics
advanced drug delivery
1. Introduction
The challenges surrounding the use of metered dose inhalers (MDIs) became a major driver for the development of dry powder formulations and dry powder inhaler (DPI) technologies [1][2]. However, the manufacture and development of these DPI products are rather complex and create challenges with respect to establishing bioequivalence (BE) between the proposed generic product and the reference listed drug (RLD), also commonly known as the brand name drug product. There are many different excipients and manufacturing methods that can be utilized to develop dry powder formulations, and each formulation requires careful study to understand how it affects product performance. Furthermore, unlike MDIs which generally follow a more standardized actuator design, there are many different DPI device types and designs. These DPI device factors also influences product performance as well as the ability for patients to utilize the device, since each DPI device can have a unique design and administration procedure.
2. Overview of DPI Products
DPIs are commonly used drug products for modern inhalation therapy of respiratory diseases that have specially designed delivery devices. These devices were developed to overcome certain limitations of MDIs, such as the complex coordination required for optimal lung delivery during times when patients may be undergoing bronchospasms [3], as well as the bulkiness and drug delivery duration of nebulizer devices. Most DPI devices are breath-actuated, which inherently avoids the need to synchronize device actuation with inspiration maneuvers.
Most DPIs contain three functional parts: powder drug formulation, drug dose measuring system, and a physical mechanism that allows dispersion of the powdered formulation.
2.1. Drug Formulation as a Powder
A DPI’s formulation is typically a complex dry powder mixture consisting of one or more active pharmaceutical ingredients (API) along with inactive, excipient ingredients. The use of excipients in the development of the formulation may not only enhance the chemical and physical stability of the API but can also impact the performance of the product. For example, this may include changing the dissolution of the API, the particle size, deposition to the lungs, and therapeutic efficacy. In general, excipients are usually “generally recognized as safe” (GRAS) substances that may improve the delivery and performance of a drug but cannot exert therapeutic effects by themselves [4]. Compared to other routes of administration, e.g., oral, topical, or parenteral, the number of excipients currently used or approved in inhalation drug products is fairly limited. The choice of excipients may depend on the disease being treated as well as the target region for delivery. Notably, the amount of the API in a single dose from a DPI is often very small, which makes dispensing a reproducible amount with each actuation challenging. Therefore, in most dry powder formulations, the excipients are used as carrier particles that provide bulk to the formulation, thereby improving the metering, dispensing, and handling of the formulation [5]. Traditional DPI formulations consist of micronized drug particles blended with lactose, a disaccharide composed of galactose and glucose, as coarse carrier particles. Lactose is an inexpensive excipient with an established stability and safety profile that is available in different grades [6][7]. Lactose is known to improve powder flowability thereby improving the reproducibility of the dose and as a diluent [8]. During drug formulation manufacture, lactose in the DPI formulation acts as a stabilizer for the spray drying process [8].
When selecting which excipient(s) to include in a DPI’s formulation, understanding how the excipient properties will influence the formulation or affect its biocompatibility with the site of action in the lungs is critical. Polymers, such as methylcellulose that are usually used in oral formulations, cannot be used as excipients in DPI formulations because these are non-degradable and, therefore, should not be delivered to the lungs. Similarly, polysorbates or oleic acid, used as excipients in MDI formulations, may not be used in DPI formulations due to their semi-solid or liquid states and low melting points which are not suitable for dry powder phase-based formulation. While lactose is a well-known DPI excipient, its powder structure may impact the inhalation efficiency of the DPI formulation. For example, amorphous lactose can give rise to strong particle–particle interactions that may impede aerosolization of the formulation [9]. To help overcome this, magnesium stearate (MgSt) can be employed as an adsorbed coating to reduce particle flocculation and thereby enhance the performance of lactose-based DPI formulations [10].
Sugars, such as mannitol and trehalose, are other excipients commonly used in DPI formulation development. Mannitol is a sugar alcohol and being a non-reducing compound, it is compatible with APIs containing amines [11]. Mannitol is also less hygroscopic than lactose and, therefore, cannot be used as a stabilizer for spray dried DPI formulations because of the rapid crystallization [12]. In addition, high amounts of mannitol can create a hyperosmolar environment and, therefore, are typically not used in DPI formulations for the management of asthma [13]. Trehalose is also a non-reducing disaccharide that may be used as a stabilizer for spray-dried inhalation powders. However, because of the hygroscopic nature of amorphous trehalose, it is often combined with other excipients, such as leucine [13].
Other prospective sugars have been evaluated, but they generally give rise to similar limitations as trehalose. For example, sorbitol, xylitol, and maltitol, have been evaluated as excipients for DPIs, but their hygroscopic nature and sensitivity to humidity have generally precluded their use in these drug products [14]. The use of amino acids such as trileucine as an excipient can improve powder dispersibility and reduce moisture uptake by the dry powder formulation. Trileucine is an efficient surface-active agent which provides a hydrophobic surface to the dry powder particles, thereby producing particles with low cohesivity, thus, improving the aerosol efficiency [15]. Fumaryl diketopiperazine (FDKP) is another excipient that is utilized with the proprietary TechnoSphere® formulation, such as found in the recently approved Tyvaso® DPI [16]. FDKP is a highly water-soluble compound that precipitates and agglomerates into low density particles under acidic conditions to form an encapsulating layer around the API [3].
2.2. Drug Delivery and the Device Landscape
There are many variations when it comes to DPI device designs, which are the result of optimization for formulation delivery and patient use. Some of the important functions of a DPI device are:
-
Ability to protect the drug formulation from environmental factors (e.g., humidity, light, dust);
-
Minimize residual drug remaining after device actuation;
-
Consistently deliver a metered dose;
-
Have a resistance appropriate to achieve the desired flow rate;
-
Enable patient compliance and be easy to use with minimal dose administration steps.
Furthermore, depending on the frequency of dosage and the API, devices will be either a “single-dose” system or a “multi-dose” system, and can come as reusable or disposable devices. The formulation and clinical application also dictate how the drug will be stored in the device to maximize the emitted dose (ED) [17].
Currently, there are three types of DPI drug storage systems—capsules, blister packages, and reservoirs. Capsules for DPIs are typically composed of gelatin or hydroxypropyl methylcellulose (HPMC) [18][19], but may be composed of different materials depending on the formulation. It is desirable for the capsule composition to be inert and not interact with the formulation as this helps in maximizing the ED. These capsules are fitted into the DPI device and pin punctured so that upon inhalation, the drug is released from the capsule and delivered to the patient. In contrast, blister packages are usually foil-based containers that are either peeled or punctured, depending on the device mechanism, to release the drug powder formulation [20][21][22][23][24][25]. Finally, reservoirs are small container fittings for the DPI which carry the drug powder formulation as a bulk product and either go through a metering system for a multi-dose device or are stored in disposable reservoirs for single-dose devices. Below is a brief examination of some examples of different DPI device designs that are either marketed in the U.S. or elsewhere or are being researched.
HandiHaler and Cyclohaler: The HandiHaler® (Figure 1) and Cyclohaler® DPI devices are classified as single-dose, capsule-based devices. Multiple drugs are delivered by the HandiHaler® and Cyclohaler® DPI devices—salmeterol xinafoate, beclomethasone dipropionate, ipratropium bromide, budesonide, formoterol fumarate, and tiotropium bromide. In the U.S., the HandiHaler® DPI device is marketed to deliver tiotropium bromide under the brand name Spiriva HandiHaler®. This drug–device product has three major steps: after the capsule has been inserted into the device, it releases a single dose upon puncture on both long ends of the capsule, and the contained powder drug is delivered by breath actuation [3]. The HandiHaler® has a slightly higher airflow resistance compared to the Cyclohaler® [26]. Typically, it is understood that a higher airflow resistance indicates the need for a higher inspiratory flow rate, while a lower airflow resistance indicates the need for a lower inspiratory flow rate; however, this is not necessarily the case always for all devices that have a high, medium, or low airflow resistance.

Figure 1. Diagram of the Spiriva HandiHaler [27].
Podhaler: The TOBI® Podhaler® (Figure 2) was approved in 2013 for the treatment of Pseudomonas infections in cystic fibrosis patients. The Podhaler® (developed by Novartis Pharmaceuticals Corporation) was seen in a more positive light than nebulizers, which were inconvenient for transport and could easily be contaminated [3][28]. The TOBI® Podhaler® also utilizes a novel porous particle formulation technology known as PulmoSphere™ which is a registered trademark of Novartis AG, licensed to the Viatris Companies. PulmoSphere™ particles have advanced aerodynamic properties to enable lung deposition of the antibiotic tobramycin. The Podhaler® device [28] is also used for the delivery of other antibiotics such as ciprofloxacin. It is a cylindrical shaped, capsule-based device which consists of a mouthpiece, chamber, body, and button to pierce the capsule so that the powder may be released upon breath actuation [3][28][29]. It is classified as a multi-unit dose device, and the formulation is phospholipid based. The Podhaler® was designed to have a low airflow resistance to allow patients to generate high airflow rates (40–85 LPM) and attain reliable dose delivery [3].

Figure 2. Diagram of the TOBI® Podhaler® [30].
The Ellipta® is a family of DPIs utilized for the treatment of chronic obstructive pulmonary disease (COPD) symptoms and asthma (Figure 3). For example, Breo Ellipta® contains double foil blister packs of fluticasone furoate and vilanterol trifenatate as maintenance therapy for COPD patients and in order to reduce exacerbations. The device comes pre-loaded with the drug blister packs and is actuated by releasing a lever on the side of the device and then inhaling through the mouthpiece. The device has medium resistance and is reported to have an intuitive device design for the patient [22][31].
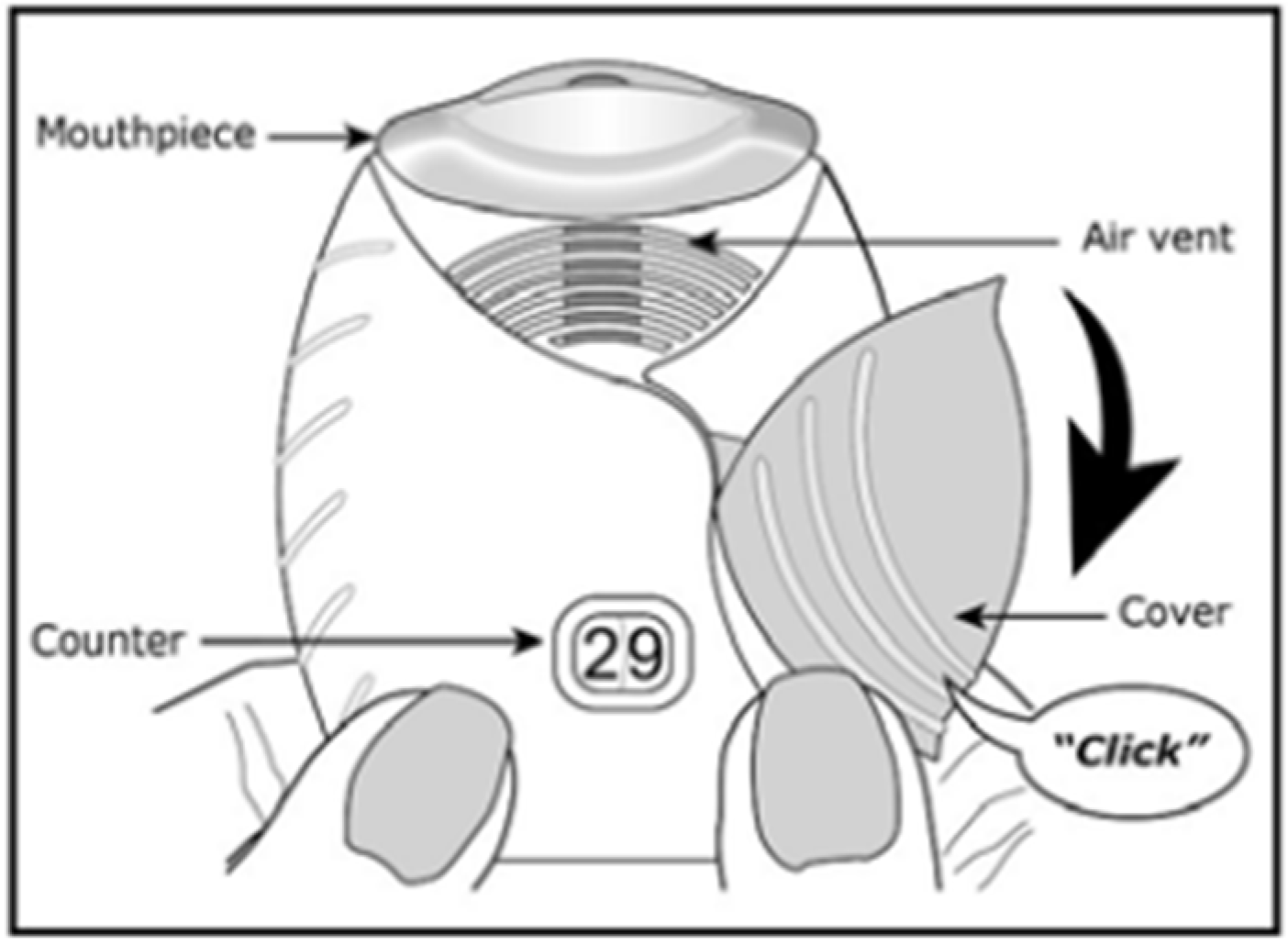
Figure 3. Diagram of Breo Ellipta® [32].
The Dreamboat is a reusable multi-unit dose, cartridge-based device [3]. It is for the systemic delivery of insulin. The insulin powder is prepared by freeze-drying with a novel excipient, FDKP, to form porous particle TechnoSphere®. The pre-metered plastic cartridges contain a dose of either 4 IU or 8 IU insulin. Once the dose has been delivered, the cartridge is removed. The inhaler deagglomerates the powder in a convergence zone where two independent flow paths intersect. The inhaler has a relatively higher resistance; however, due to the population excluding those that have pulmonary diseases, the patient should be able to generate sufficient flow to actuate the inhaler.
The Twisthaler® (Figure 4) is an example of a reservoir-based DPI for the delivery as part of Asmanex® Twisthaler® drug product that contains mometasone furoate indicated for the treatment of asthma. It is a reusable multi-dose, breath-actuated device [33][34]. Prior to inhalation, the patient must twist the mouthpiece until a click is heard to prepare the device with drug product for inhalation. The device is used for both children and adults and belongs to the class of higher-resistance devices [35].

Figure 4. Asmanex® Twisthaler® [36].
The TwinCaps® is a single-use disposable multi-unit dose inhaler, designed to be marketed as a pre-filled, low-cost inhaler to deliver large doses of drug. It was predominantly used in Japan for the systemic delivery of laninamivir (laninamivir octanoate hydrate), a novel neuraminidase inhibitor for the treatment and postexposure prophylaxis of influenza via pulmonary administration. The inhaler consists of two plastic parts: a plastic housing and a twin reservoir to carry the drug. The device has intermediate airflow resistance and is operated by sliding the reservoir chamber from side to side and inhaling. The device is primed by vertically tapping downward on a hard surface to make sure all the powder has settled to the bottom. The formulation is mixed with lactose blend for manufacturing the dry powder.
While each of these device designs comes with its benefits, it is also important to note that each design has its limitations. With capsule based DPIs that require single doses, for example, loading the capsule into the inhaler immediately before use may be a maneuver potentially inconvenient for some patients since this does not allow direct counting of the remaining doses [17]. Furthermore, properly loading the capsule-based DPIs requires a sequence of steps that may not be easy for patients with reduced dexterity. Some of these capsule-based DPIs require eight steps to inhale the medication (e.g., the Breezhaler® and HandiHaler®), and this may not be as intuitive for patients and hence contribute to reduced compliance [17][18]. On the other hand, patients may have difficulty loading and cleaning the blister-based devices. If the blister packs are not completely pierced, it may lead to incomplete drug delivery, or patients may cover the air inlet holes with their mouths while inhaling due to incorrect positioning. With reservoir devices, the drawback may come from the patient not holding the device correctly in a way that the reservoir empties the full dose.
2.3. Patient Aspects
2.3.1. Disease Conditions Treated
DPIs have been commonly used to treat patients with respiratory diseases, especially asthma and COPD. Critical to disease treatment and control is the delivery of the targeted dose from the device to the lungs. However, dose delivery is not only fulfilled by the optimal design of DPI devices but also affected by the inspiratory flow generated by the patient. This is largely determined by the severity of the patient’s disease. For example, asthma and COPD are both characterized by airway narrowing that limits air flow and gas exchange. Ultimately, the kinetic energy of the inspiratory air flow should efficiently deagglomerate dry powder and generate aerosol containing particles in the aerodynamic size range of 1–5 µm suitable for lung deposition.
Relevant to the inspiratory flow rate is the severity of disease. Here the researchers will discuss the severity classification and treatment for both diseases which are closely related to the DPI performance. The Expert Panel Report 3 (EPR 3): Guidelines for the Diagnosis and Management of Asthma (2007) provides detailed clinical information on asthma management [37]. In general, disease severity can be determined using pulmonary function testing such as spirometry, which measures the air volume that is breathed in and out of the lungs in one forced breath. A common parameter measured in spirometry, FEV1, represents the forced expiratory volume of air in one second. A predicted FEV1 value is obtained from a given healthy population, taking into consideration other factors, such as age, gender, and height. The percentage of predicted FEV1 value in an individual patient can be used for severity classification.
Asthma and COPD patients use a large variety of DPIs delivering different classes of medications for disease control. Although the treatment choice for COPD patients is typically individualized, based on a variety of factors including drug benefit/risk analysis and disease severity, these patients are often treated with long-acting muscarinic antagonist (LAMA) in combination with a long-acting beta2-antagonist (LABA). The LAMA-LABA dual treatment has been found to significantly improve symptoms and lung functions, reduce exacerbation rates, and decrease hospitalizations. Regardless, the severe impairment of lung function in severe asthma and COPD patients can pose a challenge to the targeted dose delivery for DPIs, as these patients may generate insufficient inspiratory flow for dose delivery. Moreover, device resistance is also a critical factor that affects the turbulent energy required for powder deagglomeration and dose delivery.
2.3.2. Importance of Inhalation, User Interface, and Coordination
Patient-controlled factors, such as whether a patient’s training to use the DPI product is successful, impact a patient’s inhalation technique, adherence, and, ultimately, the success of treatment outcomes. DPIs, whether breath-actuated or passive systems, were developed to address challenges of inhalation and coordination that patients face when using powder MDIs and are generally considered to minimize the patient–device coordination [38][39][40]. Passive DPIs allow for the patient to coordinate the actuation process by using their own breath to initiate and complete dose delivery from the device. While this process eases the coordination between actuation and inhalation steps, the use of passive DPIs still require a sufficient inhalation technique to achieve the desired delivered dose to the target regions within the lung [41]. This is critical as the peak inhalation flow (PIF) achieved by patients through each DPI product has been shown to be tied to the clinical efficacy [42]. A proper inhalation technique from a patient does rely on successful patient training by a healthcare provider, which is recommended in clinical practice guidelines [43]. However, in the real word, both patients and physicians struggle to master the proper inhalation technique due to a myriad of factors. Some patient populations may struggle in understanding critical steps for DPI use, may be physically or cognitively limited due to age (most notably children and the elderly) or lack of education, or may receive insufficient training from their healthcare professional [34][37]. Healthcare professionals may also struggle with training by failing to understand the proper DPI device technique, lack of time for appropriate review of the patient’s inhalation technique, or due to poor communication with the patient [32][34][37]. The lack of confidence in the proper inhalation technique felt by a patient can lead to nonadherence and poor disease control exacerbations, hospitalizations, and, in some cases, the need for oral medications to control the patient’s asthma or COPD condition [34][37][44]. Thus, proper education, communication, and training regarding the patient and healthcare level are simple steps that are likely to improve therapeutic outcomes.
While proper education and training may address some concerns, other confounding factors that impact proper inhalation technique and clinical outcomes are related to the DPI device design and the variety of DPI device types that exist on the market. Passive DPIs utilize the patient’s inhalation as the energy source to achieve the ED from the device. Each DPI device design has a unique internal geometry, airflow path, and other internal features to assist in the deagglomeration process of the API from their carrier particles and achieve a desired aerosol performance [35]. Since each DPI design uses a unique dispersion process and/or internal geometry and airflow pathway, the internal resistance to airflow is also unique to each DPI device design [35][45]. The correct inhalation technique is clinically important because these devices utilize the interaction between the patient’s inhalation flow and the internal resistance of the inhaler to generate the turbulent airflow energy necessary to deagglomeration and aerosolize the powder formulation and achieve the desired respirable dose [36][46][47][48][49]. Thus, the patient’s inhalation effort and the device’s internal resistance to airflow are critical to achieve the desired lung dose.
When developing a generic DPI, an emphasis on the device design is expected as the DPI device and its interaction with the formulation and patient’s ability to properly use the device are essential to achieve an optimized aerosol performance [35]. Therefore, a generic device should be substitutable to the RLD DPI device when used by the patient. Specifically, a generic DPI is expected to contain similar external operating principles and external critical design attributes (e.g., size, shape, and operating steps) as the brand (RLD) product it intends to reference. The generic DPI device is also expected to contain the same metering principle to the RLD DPI device (e.g., a metered multi-dose format for reservoir or blister based DPIs, a pre-metered single-unit dose capsule-based format for capsule based DPIs).
The user interface encompasses the external critical design attributes, those aspects of the device related to how a patient uses the product for drug administration [45]. The user interface includes all components of a drug–device combination product that a user interacts with, including the delivery device, associated device controls and displays, product labeling, and product packaging. When developing a generic DPI product, the generic manufacturer is expected to assess the user interface of their proposed product as described in the draft FDA guidance for industry, Comparative Analyses and Related Comparative Use Human Factors Studies for a Drug-Device Combination Product Submitted in an ANDA [43]. This outlines the U.S. Food and Drug Administration’s (FDA) current thinking on what and how to compare the user interface of a proposed generic to its intended RLD product. As explained in this guidance, the assessment includes a labeling comparison, a comparative task analysis, and a physical comparison of the delivery device constituent part to analyze any potential differences (e.g., no design differences, minor design differences, or other design differences) between the user interface of the generic product and RLD product [43].
Overall, the design of the user interface for a generic DPI is not expected be identical to its RLD, but it is expected to produce the same clinical effect and safety profile as the RLD under the conditions specified in the labeling without the intervention of a health care provider and/or without additional training prior to use. Generally, any differences in user interfaces should be adequately analyzed and scientifically justified via a comparative (threshold) analyses and, if necessary, additional data such as a comparative human factors study. Considerations, such as the indication, context of use of the product (emergency vs. non-emergency use, daily vs. intermittent use), the end user (pediatric and/or adult patients vs. health care provider), potential use errors, and user error risks are considered during the user interface assessment of any generic and RLD DPIs. Thus, it is critical to consider these aspects when developing a generic drug–device combination product.
2.4. General BE Recommendations
To establish that a proposed generic drug product is bioequivalent to its RLD, the generic drug applicant must demonstrate that there is no significant difference in the rate and extent of the API (in pharmaceutical equivalents) becoming available at the site of action as the reference standard when the same molar dose is administered in an adequately designed study. As detailed above, the performance of inhalation drug products like DPIs can be influenced by many different factors, such as the various aspects of the formulation, its interaction with the device characteristics, and the disease condition being treated. With such a varied number of influencing factors, DPIs are considered complex drug–device combination products that require a multi-faceted approach to demonstrate that a generic company’s DPI is bioequivalent to the RLD. The FDA has generally referred to this approach as the agglomeration weight of evidence approach as it includes a combination of in vitro and in vivo studies to evaluate whether differences exist in product performance, systemic exposure, and local drug delivery between a generic product and its RLD [50]. The FDA has published product-specific guidances (PSGs) that outline the FDA’s recommendations for demonstrating BE for a number of DPI products [51].
For DPIs, the evaluation of product performance is conducted through in vitro BE studies that measure single actuation content (SAC) and aerodynamic particle size distribution (APSD). In general, in vitro BE studies are sensitive to detecting differences in formulation, device, and manufacturing process that could impact product performance. For DPI products, both SAC and APSD studies have been recommended as they are believed to be relevant to the lung regional and total drug deposition performance for these products. Importantly, these studies are also conducted across a range of airflow rates that encompass the inspiratory flow ranges of the indicated patient population, thereby ensuring that any variability in a patient’s ability to inhale through the proposed generic DPI will not result in performance differences that may affect its BE to the RLD.
The in vivo BE studies typically recommended for DPIs include both pharmacokinetic (PK) studies and comparative clinical endpoint or pharmacodynamic (PD) studies. PK BE studies are included in the weight of evidence approach to evaluate whether there is a difference in the systemic exposure of the generic product and RLD that may lead to differences in side effects or any adverse reactions. It is recommended that these studies use a single-dose design in healthy volunteers instead of patients, given that healthy volunteers generally provide a lower PK variability since patient-related factors, such as disease severity, are not present. As with the in vitro BE studies, PK BE studies for DPIs are recommended for each strength, using a dose that, while based on the analytical assay sensitivity, requires a minimum number of inhalations. For establishing BE in local drug delivery, comparative clinical endpoints or PD BE studies are recommended to be conducted in one of the indicated patient populations using the lowest dose listed in the approved drug label. Comparative clinical endpoints or PD BE studies are often considered the most challenging studies for generic applicants to complete since these studies are generally considered to be less sensitive in detecting formulation difference as compared to other methods. Their use of the more variable patient population as well as the challenges stemming from the available endpoints for measuring local treatment effects often requires significant numbers of patients in each treatment arm to achieve sufficient statistical power for evaluating BE.
In addition to the in vitro and in vivo BE studies described above, the FDA generally recommends that a generic DPI product be formulated qualitatively (Q1) and quantitatively (Q2) in the same way as the RLD product and have device similarity,. With that said, it is also important to note that, as per the U.S. Code of Federal Regulations (CFR), DPI generic products are not required to be formulated the same as their respective RLD product. Therefore, the FDA’s BE recommendations include approaches for generic DPI formulations with greater than 5% differences in an excipient amount compared to the RLD, so long as the generic applicant can provide adequate justification that this difference does not affect the product’s safety or efficacy. This may include in vitro testing across multiple drug-to-excipient ratios. In general, inclusion of formulation sameness criteria as part of a BE recommendation reduces the likelihood that a generic product will not be bioequivalent to the RLD. In addition, the criteria reduce the chance that differences in the excipients may lead to changes in safety of the product.
3. Advancements in DPI Device Technologies
In addition to the advancements in DPI formulation design, advancements are also continuing on the device front with the introduction of new designs and digital technologies. With the inclusion of digital technology, these digital DPI platforms have the ability to provide additional information on inhaler performance and/or use that may minimize device use errors. This has shown a positive impact on product use and performance, as well as patient satisfaction, compliance, and adherence, thus, overall clinical outcomes [52]. A recent review by Xiroudaki et al. 2021 summarizes the current landscape of digital DPIs [53].
The currently FDA-approved digital DPIs include the Digihaler® used with ProAir®, AirDuo®, and Armonair® products, which were approved in 2018, 2019, and 2020, respectively [53]. The Digihaler® contains an integrated digital sensor encompassed in an electronic module (eModule) on the top portion of the inhaler that can record usage (date and time), inhalation data relevant to the inhalation technique (e.g., PIF and flow volume), and provide medication reminders [53][54]. The accuracy, benefits, and outcomes of such digital DPIs, such as the Digihaler®, are beginning to emerge [55][56][57]. Other FDA-approved digital DPIs included add-on sensors rather than being fully integrated in the DPI device. The Propeller® sensors designed for several DPI devices including Diskus® (GlaxoSmithKline, Brentford, UK), Ellipta (GlaxoSmithKline, Brentford, UK), and Neohaler (Novartis, Basel, Switzerland) were approved by the FDA through the 510(k) pathway in 2015, 2016, and 2018, respectively [53]. The Propeller® sensors technology can record and monitor actuation activity (date and time) and utilizes Global Positioning System (GPS) technology to identify environmental asthma exacerbation triggers. Additionally, the Hailie® sensor (approved via the 510(k) pathway) was developed for MDIs as well as the Diskus® (SmartDisk™) and HandiHaler® (SmartHandy™), which is capable of being attached to the inhaler to record and track medication usage and set reminders.
As the inclusion of digital sensors to DPIs (either added on or fully integrated) become more commonplace, the real-world evidence of their potential benefits may begin to show, such as changes in patient adherence and clinical outcomes. As mentioned earlier, other inhalation products such as MDIs and nebulizer-based products can present challenges to patient adherence and outcomes due to their need for better coordination with the patient, or the potential device bulkiness and long duration of administration. While DPIs can address these challenges due to their passive drug delivery approach and size, the cost for patients to use a DPI over other inhalation products may also be a consideration that impacts patient adherence and clinical outcomes. So, while the addition of digital versions of DPIs may offer benefits to patients, these would need to be considered along with any differences in costs for the patient when attempting to understand potential changes to patient behavior such as adherence to a prescribed treatment. Currently, there are no approved generic DPIs that incorporate a digital feature. These digital technologies present a new challenge for generic DPI development given the uncertainty with degree to which a generic DPI will be required to include digital technologies, or record and/or communicate this information to the patient, in order to match the RLD DPI. With that said, generic competition with these DPIs is expected to lead to reduced costs for the patient, which has been reported following the introduction of the first Advair Diskus generic [58]. As this new area continues to develop and bring with it new regulatory challenges, the FDA continues to evaluate these challenges through regulatory science initiatives that will aid future generic development.
Lastly, the introduction of the Staccato inhalation platform provides an example of the continued device design advancements for inhalation powders. The Staccato device stands out among other inhalation devices due to its drug delivery mechanism. The device uses breath actuation, where an inhalation sensor triggers a heating element on the device that heats up a thin film of the formulation, causing it to sublimate and aerosolize the dose as dry particles [59]. Notably, the Staccato device’s use of a thin film formulation makes it distinct from other carrier-based DPI formulations as the formulation is not in a powder form prior to actuation. Currently, the Staccato device is used in the FDA-approved product Adasuve (loxapine) inhalation powder.
References
- De Boer, A.H.; Hagedoorn, P.; Hoppentocht, M.; Buttini, F.; Grasmeijer, F.; Frijlink, H.W. Dry powder inhalation: Past, present and future. Expert Opin. Drug Deliv. 2017, 14, 499–512.
- Stein, S.W.; Thiel, C.G. The history of therapeutic aerosols: A chronological review. J. Aerosol Med. Pulm. Drug Deliv. 2017, 30, 20–41.
- Berkenfeld, K.; Lamprecht, A.; McConville, J.T. Devices for dry powder drug delivery to the lung. AAPS Pharmscitech. 2015, 16, 479–490.
- Pilcer, G.; Amighi, K. Formulation strategy and use of excipients in pulmonary drug delivery. Int. J. Pharm. 2010, 392, 1–19.
- Feeley, J.C.; York, P.; Sumby, B.S.; Dicks, H. Determination of surface properties and flow characteristics of salbutamol sulphate, before and after micronisation. Int. J. Pharm. 1998, 172, 89–96.
- Telko, M.J.; Hickey, A.J. Dry powder inhaler formulation. Respir. Care 2005, 50, 1209–1227.
- Smyth, H.D.; Hickey, A.J. Carriers in drug powder delivery. Am. J. Drug Deliv. 2005, 3, 117–132.
- Healy, A.M.; Amaro, M.I.; Paluch, K.J.; Tajber, L. Dry powders for oral inhalation free of lactose carrier particles. Adv. Drug Deliv. Rev. 2014, 75, 32–52.
- Steckel, H.; Bolzen, N. Alternative sugars as potential carriers for dry powder inhalations. Int. J. Pharm. 2004, 270, 297–306.
- Jetzer, M.W.; Schneider, M.; Morrical, B.D.; Imanidis, G. Investigations on the mechanism of magnesium stearate to modify aerosol performance in dry powder inhaled formulations. J. Pharm. Sci. 2018, 107, 984–998.
- Mensink, M.A.; Frijlink, H.W.; van der Voort Maarschalk, K.; Hinrichs, W.L. How sugars protect proteins in the solid state and during drying (review): Mechanisms of stabilization in relation to stress conditions. Eur. J. Pharm. Biopharm. 2017, 114, 288–295.
- Hertel, N.; Birk, G.; Scherließ, R. Particle engineered mannitol for carrier-based inhalation–a serious alternative? Int. J. Pharm. 2020, 577, 118901.
- Zillen, D.; Beugeling, M.; Hinrichs, W.L.; Frijlink, H.W.; Grasmeijer, F. Natural and bioinspired excipients for dry powder inhalation formulations. Curr. Opin. Colloid Interface Sci. 2020, 56, 101497.
- Tee, S.K.; Marriott, C.; Zeng, X.M.; Martin, G.P. The use of different sugars as fine and coarse carriers for aerosolised salbutamol sulphate. Int. J. Pharm. 2000, 208, 111–123.
- Lechuga-Ballesteros, D.; Charan, C.; Stults, C.L.; Stevenson, C.L.; Miller, D.P.; Vehring, R.; Kuo, M.C. Trileucine improves aerosol performance and stability of spray-dried powders for inhalation. J. Pharm. Sci. 2008, 97, 287–302.
- Angelo, R.; Rousseau, K.; Grant, M.; Leone-Bay, A.; Richardson, P. Technosphere® insulin: Defining the role of technosphere particles at the cellular level. J. Diabetes Sci. Technol. 2009, 3, 545–554.
- Rossi, I.; Ganley, W.J.; Michelet, O.; Grosjean, B.; Sarrailh, S.; Williams, G.; Price, R.; Shur, J. The role of in silico regional deposition modelling and pharmacokinetic profiling in the development of a generic tiotropium dry powder inhaler. Respir. Drug Deliv. 2020, 393–396. Available online: https://nanopharm.co.uk/wp-content/uploads/2020/08/Rossi-2020b.pdf (accessed on 24 October 2022).
- Lavorini, F.; Pistolesi, M.; Usmani, O.S. Recent advances in capsule-based dry powder inhaler technology. Multidiscip. Respir. Med. 2017, 12, 1–7.
- Buttini, F.; Quarta, E.; Allegrini, C.; Lavorini, F. Understanding the Importance of Capsules in Dry Powder Inhalers. Pharmaceutics 2021, 13, 1936.
- Donovan, M.J.; Gibbons, A.; Herpin, M.J.; Marek, S.; McGill, S.L.; Smyth, H.D. Novel dry powder inhaler particle-dispersion systems. Ther. Deliv. 2011, 2, 1295–1311.
- Ninane, V.; Vandevoorde, J.; Cataldo, D.; Derom, E.; Liistro, G.; Munghen, E.; Vincken, W. New developments in inhaler devices within pharmaceutical companies: A systematic review of the impact on clinical outcomes and patient preferences. Respir. Med. 2015, 109, 1430–1438.
- Grant, A.C.; Walker, R.; Hamilton, M.; Garrill, K. The ELLIPTA® dry powder inhaler: Design, functionality, in vitro dosing performance and critical task compliance by patients and caregivers. J. Aerosol Med. Pulm. Drug Deliv. 2015, 28, 474–485.
- Chrystyn, H. The DiskusTM: A review of its position among dry powder inhaler devices. Int. J. Clin. Pract. 2007, 61, 1022–1036.
- Laube, B.L. The expanding role of aerosols in systemic drug delivery, gene therapy, and vaccination. Respir. Care 2005, 50, 1161–1176.
- Krueger, M.; Metzger, B.; Trunk, M.; Schiewe, J. Boehringer Ingelheim International GmbH, assignee. Blister for inhalers. U.S. Patent Application US 12/949,401, 17 March 2011.
- Shur, J.; Lee, S.; Adams, W.; Lionberger, R.; Tibbatts, J.; Price, R. Effect of device design on the in vitro performance and comparability for capsule-based dry powder inhalers. AAPS J. 2012, 14, 667–676.
- , NDA 021395. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/021395s048lbl.pdf (accessed on 24 October 2022).
- VanDevanter, D.R.; Geller, D.E. Tobramycin administered by the TOBI® Podhaler® for persons with cystic fibrosis: A review. Med. Devices 2011, 4, 179.
- Geller, D.E.; Weers, J.; Heuerding, S. Development of an inhaled dry-powder formulation of tobramycin using PulmoSphere™ technology. J. Aerosol Med. Pulm. Drug Deliv. 2011, 24, 175–182.
- , NDA 201688. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/201688s010lbl.pdf (accessed on 24 October 2022).
- Canadian Agency for Drugs and Technologies in Health. Fluticasone Furoate/Vilanterol (Breo Ellipta). 2016. Available online: https://www.ncbi.nlm.nih.gov/books/NBK409785/ (accessed on 24 October 2022).
- , NDA 204275. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/204275s017lbl.pdf (accessed on 24 October 2022).
- Asmanex Twisthaler. Available online: https://www.asmanex.com/asmanex-twisthaler/ (accessed on 24 October 2022).
- Nationwide Children’s Hospital. Available online: https://www.nationwidechildrens.org/family-resources-education/health-wellness-and-safety-resources/resources-for-parents-and-kids/how-to-use-an-epipen/epinephrine-myths-and-facts/how-to-use-a-twisthaler#:~:text=A%20twisthaler%20is%20a%20dry,delivering%20a%20puff%20of%20medicine (accessed on 24 October 2022).
- Dal Negro, R.W. Dry powder inhalers and the right things to remember: A concept review. Multidiscip. Respir. Med. 2015, 10, 1–4.
- , NDA 021067. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/021067s032lbl.pdf (accessed on 24 October 2022).
- Expert Panel Report 3: Guidelines for the Diagnosis and Management of Asthma. Available online: https://www.epa.gov/asthma/expert-panel-report-3-guidelines-diagnosis-and-management-asthma (accessed on 24 October 2022).
- Dolovich, M.B.; Dhand, R. Aerosol drug delivery: Developments in device design and clinical use. Lancet 2011, 377, 1032–1045.
- Khassawneh, B.Y.; Al-Ali, M.K.; Alzoubi, K.H.; Batarseh, M.Z.; Al-Safi, S.A.; Sharara, A.M.; Alnasr, H.M. Handling of inhaler devices in actual pulmonary practice: Metered-dose inhaler versus dry powder inhalers. Respir. Care 2008, 53, 324–328.
- Lavorini, F.; Magnan, A.; Dubus, J.C.; Voshaar, T.; Corbetta, L.; Broeders, M.; Crompton, G.K. Effect of incorrect use of dry powder inhalers on management of patients with asthma and COPD. Respir. Med. 2008, 102, 593–604.
- Newman, B.; Witzmann, K. Addressing the regulatory and scientific challenges with generic orally inhaled drug products. Pharm. Med. 2020, 34, 93–102.
- Azouz, W.; Chetcuti, P.; Hosker, H.S.; Saralaya, D.; Stephenson, J.; Chrystyn, H. The inhalation characteristics of patients when they use different dry powder inhalers. J. Aerosol Med. Pulm. Drug Deliv. 2015, 28, 35–42.
- Plaza, V.; Giner, J.; Rodrigo, G.J.; Dolovich, M.B.; Sanchis, J. Errors in the use of inhalers by health care professionals: A systematic review. J. Allergy Clin. Immunol. Pract. 2018, 6, 987–995.
- Molimard, M.; Raherison, C.; Lignot, S.; Depont, F.; Abouelfath, A.; Moore, N. Assessment of handling of inhaler devices in real life: An observational study in 3811 patients in primary care. J. Aerosol Med. 2003, 16, 249–254.
- Donovan, M.J.; Kim, S.H.; Raman, V.; Smyth, H.D. Dry powder inhaler device influence on carrier particle performance. J. Pharm. Sci. 2012, 101, 1097–1107.
- Chrystyn, H.; Lavorini, F. The dry powder inhaler features of the Easyhaler that benefit the management of patients. Expert Rev. Respir. Med. 2020, 14, 345–351.
- Clark, A.R.; Hollingworth, A.M. The relationship between powder inhaler resistance and peak inspiratory conditions in healthy volunteers—Implications for in vitro testing. J. Aerosol Med. 1993, 6, 99–110.
- Azouza, W.; Chrystyn, H. Clarifying the dilemmas about inhalation techniques for dry powder inhalers: Integrating science with clinical practice. Prim. Care Respir. J. 2012, 21, 208–213.
- Levy, M.L.; Carroll, W.; Alonso, J.L.I.; Keller, C.; Lavorini, F.; Lehtimäki, L. Understanding dry powder inhalers: Key technical and patient preference attributes. Adv. Ther. 2019, 36, 2547–2557.
- Lu, D.; Lee, S.L.; Lionberger, R.A.; Choi, S.; Adams, W.; Caramenico, H.N.; Li, B.V. International guidelines for bioequivalence of locally acting orally inhaled drug products: Similarities and differences. AAPS J. 2015, 17, 546–557.
- For More Information on the Product-Specific Guidances (PSGs) Published by the FDA. Available online: https://www.accessdata.fda.gov/scripts/cder/psg/index.cfm (accessed on 24 October 2022).
- Xiroudaki, S.; Schoubben, A.; Giovagnoli, S.; Rekkas, D.M. Dry powder inhalers in the digitalization era: Current status and future perspectives. Pharmaceutics 2021, 13, 1455.
- Price, R.; Farias, G.; Ganley, W.; Shur, J. Challenging the bioequivalence hurdles for OINDPs: Achieving Q3 structural equivalence. Respir. Drug Deliv. Asia 2018, 2018, 1–14.
- Chrystyn, H.; Saralaya, D.; Shenoy, A.; Toor, S.; Kastango, K.; Calderon, E.; Safioti, G. Investigating the accuracy of the Digihaler, a new electronic multidose dry-powder inhaler, in measuring inhalation parameters. J. Aerosol Med. Pulm. Drug Deliv. 2022, 35, 166–177.
- Mangal, S.; Conti, D.S.; Delvadia, R.R.; Oguntimein, O.M.; Shur, J.; Price, R.; Mangal, S. Microstructural Mapping of Dry Powder Inhalers (DPIs) using Morphologically Directed Raman Spectroscopy (MDRS): A novel analytical tool for DPI characterization. In AAPS PharmSci360; Walter, E., Ed.; Washington Convention Center: Washington, DC, USA, 2018.
- Chrystyn, H.; Safioti, G.; Buck, D.; Granovsky, L.; Calderon, E.; Li, T.; Pleasants, R. Real-life inhaler technique in asthma patients using the electronic ProAir Digihaler. Eur. Respir. J. 2019, 54, PA4258.
- Pleasants, R.; Safioti, G.; Reich, M.; Granovsky, L.; Itzahary, O.; Hadar, Y.; Chrystyn, H. D202 rescue medication use and inhalation patterns during asthma exacerbations recorded by Digihaler. Ann. Allergy Asthma Immunol. 2019, 123, S15.
- Wang, Z.; Ahluwalia, S.K.; Newman, B.; Dhapare, S.; Zhao, L.; Luke, M.C. Medication Cost-Savings and Utilization of Generic Inhaled Corticosteroid (ICS) and Long-Acting Beta-Agonist (LABA) Drug Products in the USA. Ther. Innov. Regul. Sci. 2022, 56, 346–357.
- Noymer, P.; Myers, D.; Glazer, M.; Fishman, R.S.; Cassella, J.V. The Staccato system: Inhaler design characteristics for rapid treatment of CNS disorders. Respir. Drug Deliv. 2010, 1, 11–20.
More
Information
Subjects:
Respiratory System
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.2K
Revisions:
2 times
(View History)
Update Date:
24 Nov 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No