Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Pietro Tedesco | -- | 1859 | 2022-11-23 11:59:08 | | | |
| 2 | Sirius Huang | Meta information modification | 1859 | 2022-11-25 01:39:24 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Dell’anno, F.; Tedesco, P.; Pascale, D.D.; Sala, G.D. Pyoverdine Biosynthesis. Encyclopedia. Available online: https://encyclopedia.pub/entry/36110 (accessed on 26 July 2026).
Dell’anno F, Tedesco P, Pascale DD, Sala GD. Pyoverdine Biosynthesis. Encyclopedia. Available at: https://encyclopedia.pub/entry/36110. Accessed July 26, 2026.
Dell’anno, Filippo, Pietro Tedesco, Donatella De Pascale, Gerardo Della Sala. "Pyoverdine Biosynthesis" Encyclopedia, https://encyclopedia.pub/entry/36110 (accessed July 26, 2026).
Dell’anno, F., Tedesco, P., Pascale, D.D., & Sala, G.D. (2022, November 23). Pyoverdine Biosynthesis. In Encyclopedia. https://encyclopedia.pub/entry/36110
Dell’anno, Filippo, et al. "Pyoverdine Biosynthesis." Encyclopedia. Web. 23 November, 2022.
Copy Citation
Pyoverdines (PVDs) are a class of siderophores produced mostly by members of the genus Pseudomonas. Their primary function is to accumulate, mobilize, and transport iron necessary for cell metabolism. Moreover, PVDs also play a crucial role in microbes’ survival by mediating biofilm formation and virulence.
pyoverdine
siderophore
iron
Pseudomonas sp.
1. Pyoverdines’ General Features
Pyoverdines (PVDs) are fluorescent molecules produced by bacteria belonging to the genus Pseudomonas (Figure 1I). Also known during the last decades as fluoresceins and pseudobactins, they were discovered at the end of the nineteenth century [1]. However, the understanding of their biological functions in microbial metabolism remained unknown until 1978, the year in which Meyer and Abdallah [2] and Meyer and Hornsperger [3] unraveled their role in the acquisition of iron. Therefore, from a functional point of view, PVDs belong to the class of siderophores, small molecules with a mass between 200 and 2000 Da that are able to chelate iron and other metals [4].

Figure 1. Structure of pyoverdine type 1 produced by P. aeruginosa (I); examples of different PVDs’ chromophore structures (II); illustration of different side chains characteristic of PVDs (III).
Iron is a crucial element for the metabolism and survival of microorganisms as it is involved in mechanisms such as the reduction of oxygen for the synthesis of ATP and DNA precursors [5][6]. Despite iron being one of the most widespread elements on our planet, its bioavailability is greatly reduced due to the reduced solubility of Fe (III) oxyhydroxide particulates, the prevailing form at neutral pH and in oxygenated environments [7].
Therefore, PVDs, and more generally siderophores, being very soluble molecules and having a high affinity with iron, exert their main role by taking up the iron present in the extracellular environment to internalize it.
From the literature, five classes of siderophores are known, each of which differs from the functional group used to bind the metals: catecholate, hydroxamate, salicylate, carboxylate, and mixed-type (Table 1).
Table 1. Representative list of siderophores belonging to the class of hydroxamate, catecholate, carboxylate, and mixed ligands.
| Type of Siderophore | Structure | Organism | References |
|---|---|---|---|
| Hydroxamate Siderophores | |||
| Albomycins | 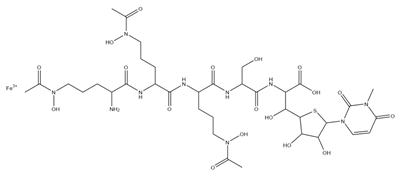 |
Actinomyces suibtropicus | [8] |
| Alcaligin | 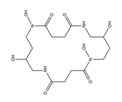 |
Bordetella pertussis; Bordetella bronchiseptica |
[9] |
| Bisucaberin | 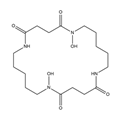 |
Alteromonas haloplanktis SB-112 | [10] |
| Coprogen | 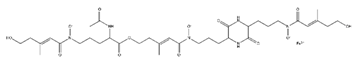 |
Trichoderm hypoxylon | [11] |
| Ferrichrome | 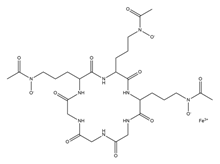 |
Lactobacillus casei | [12] |
| Ferricrocin | 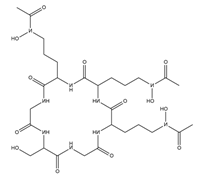 |
Trichoderma virens | [13] |
| Danoxamine | 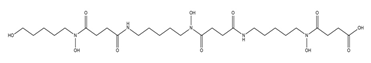 |
Streptomyces violaceus DSM 8286 | [14] |
| Deferoxamine B | 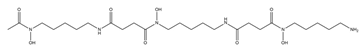 |
Streptomyces pilosus | [15] |
| Desferrioxamine E (nocardamine) |
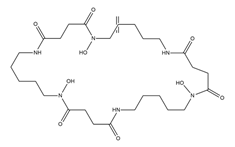 |
Streptomyces griseus | [16] |
| Fusarinine C | 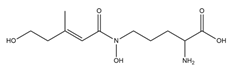 |
Fusarium roseum | [17] |
| Ornibactin | 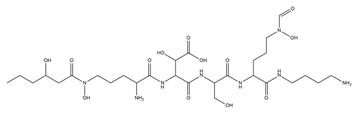 |
Burkholderia cepacia | [18] |
| Rhodotorulic acid | 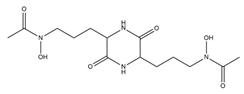 |
Rhodotorula pilimanae | [19] |
| Catecholate Siderophores | |||
| Azotochelin | 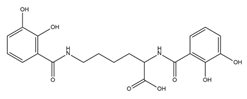 |
Azotobacter vinelandi | [20] |
| Bacillibactin | 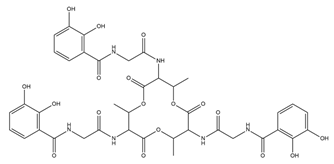 |
Bacillus subtilis, Corynebacterium glutamicum | [21] |
| Enterobactin | 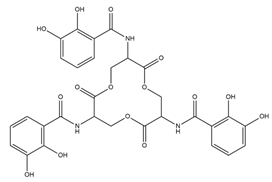 |
Escherichia coli | [22] |
| Paenibactin | 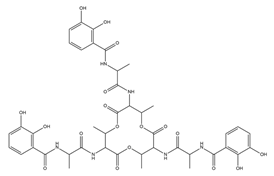 |
Paenibacillus elgii B69 | [23] |
| Protochelin | 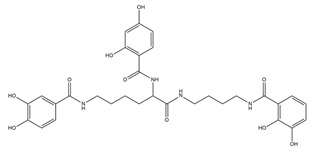 |
Azotobacter vinelandi | [24] |
| Salmochelin | 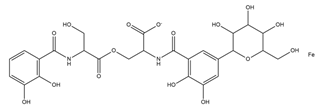 |
Salmonella enterica | [25] |
| Vibriobactin | 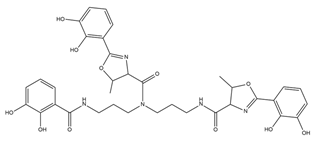 |
Vibrio cholerae | [26] |
| Carboxylate Siderophore | |||
| Achromobactin | 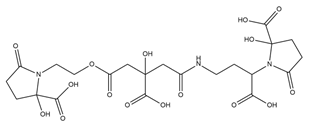 |
Erwinia chrysanthemi | [27] |
| Rhizobactin | 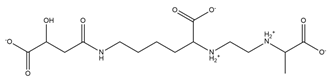 |
Rhizobium meliloti | [28] |
| Rhizoferrin | 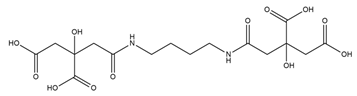 |
Rhizopus microsporus | [29] |
| Staphyloferrin A | 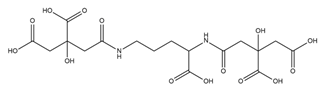 |
Staphylococcus hyicus DSM20459 | [30] |
| Mixed Ligands | |||
| Aereobactin | 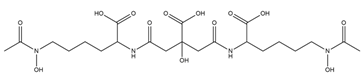 |
E. coli | [31] |
| Amychelin | 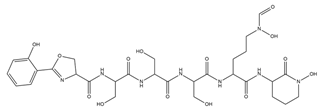 |
Amycolatopsis sp. AA4 | [32] |
| Azotobactin | 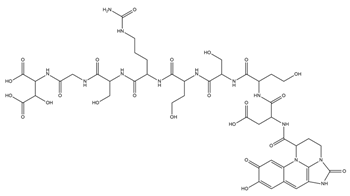 |
Azotobacter vinelandii | [33] |
| Gobichelin A and B | 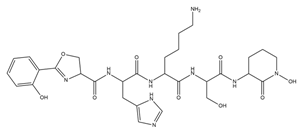 |
Streptomyces sp. NRRL F-4415 | [34] |
| Mycobactins | 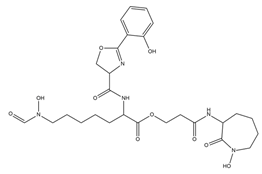 |
Mycobacterium tuberculosis | [35] |
| Pseudochelin A | 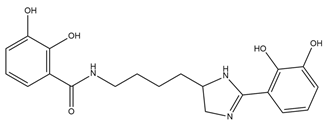 |
Pseudoalteromonas piscicida S2040 | [36] |
| Pyoverdine | 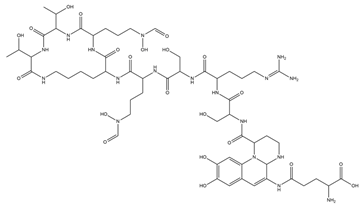 |
Pseudomonas aeruginosa | [37] |
| Rhodobactin | 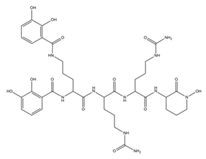 |
Rhodococcus rhodochrous strain OFS | [38] |
| Yersiniabactin | 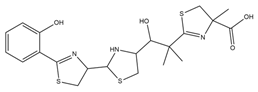 |
Yersinia pestis | [39] |
The three most represented groups of siderophores are catecholates, carboxylates, and hydroxamates [40].
PVDs belong to the mixed-type category, and like many other siderophores, are non-ribosomal peptides. Such molecules originate from non-ribosomal peptide synthetases (NRPSs), large multi-modular enzymes capable of catalysing the synthesis of peptides without an RNA template [41]. The organization in modules of the NRPSs generally contributes to the possibility of producing a wide spectrum of bioactive compounds [42] and most likely influences the specific species diversity of the different PVDs as each one is characterized by a unique peptide moiety [43]. Such a feature enables each Pseudomonas strain to acquire iron through the PVDs released by itself, or at least through heterologous PVDs sharing high homology with the native PVD [44][45], since all PVDs, once the iron is bound, only bind to a specific receptor (ferripyoverdine, FpvA) placed on the outer bacterial membrane [46]. The chemical structure of PVDs can be summarized in three parts: (i) the chromophore core (Figure 1II); (ii) the peptide backbone; and (iii) the side chain (Figure 1III). The most conserved structural element in PVDs is the chromophore, a 1S)-5-amino-2,3-dihydro-8,9-dihydroxy-1H-pyrimido-[1,2-a] quinolone-1-carboxylic acid (Figure 1III), with an absorbance at 400 nm (at neutral pH) and an emission of fluorescence at 447 nm.
However, some biological precursors and derivatives display structural modifications, such as isopyoverdine (IsoPy), dihydro-pyoverdine (DiHPy), dihyodro-pyover-dine-7-sulfonics acid (SPy), ferribactin (FerB), Azotobactin (AzoB), and succinopyoverdine (SuccPy) [47].
The other two structural features of PVDs include a variable acyl side chain attached to the 3- amino group of the fluorophore, and a strain-specific peptide backbone, usually bound to the C1 -carboxyl group of the ring system. Interestingly, the strain-specific peptide backbone varies in its sequence and can either be linear or (partially) intramolecularly cyclized [48].
Notably, the PVDs’ peptide chain is fundamental to bind iron. Indeed, PVDs chelate Fe (III) due to three bidentate chelating residues: (i) a catechol group; (ii) a hydroxamate group at the end of the peptide chain; and (iii) an hydroxamate group in the middle of the peptide chain [49][50].
Overall, the PVDs’ great affinity for iron and, more generally, metals aroused great interest among the scientific community due to their promising application in the field of human health and environment care. The main steps of PVD biosynthesis will be discussed in the following section.
2. PVD Biosynthesis
Generally, the fluorescent Pseudomonas species produce PVDs as major siderophores to access iron. In particular, P. aeruginosa strains produce three different PVDs (PVDI, PVDII, and PVDIII), each one characterized by a different peptide chain [51].
During the past few years, many reviews investigating PVD biosynthesis have been published [43][48][52][53][54][55]. This section describes, in a general way, the role of the different enzymes involved in the biosynthesis of PVD type 1 (PVDI) produced by P. aeruginosa, focusing on the most recent data of the last two years.
As stated above, PVDs are synthesized by NPRSs organized into modules (from a minimum of 2 to a maximum of 18), each of which selects and catalyzes the addition of a specific substrate to the final product. The mechanism that allows the elongation of the nascent chain is based on the presence, in each module, of three core domains: (i) adenylation (A) domain, which selects a cognate amino acid and converts it to the corresponding aminoacyl adenylate; (ii) peptidyl carrier protein (PCP) domain (also known as thiolation domain), carrying the growing peptidyl chain via a phosphopantetheinyl arm; (iii) condensation (C) domain, which catalyses the peptide bond formation [56]. The complete peptide chain is then released from the multi-enzymatic complex following the action of the thioesterase domain, present only in the termination module [57].
PVD biogenesis starts in the cytoplasm, where its biological precursor, i.e., an acylated ferribactin, is assembled by three multimodular NRPS (PvdL, PvdI, and PvdD) together with several tailoring enzymes. The four-module NRPS PvdL catalyses the biosynthesis of the chromophore precursor. The first module of PvdL recruits myristic or myristiloeic acid as a starter unit, which, as reported by [58], is supposed to prevent the diffusion of the peptide beyond the membrane during assembly. The fatty acid unit is then transferred to the second module of PvdL, catalysing the acylation of an L-glutamate residue. The next two PvdL modules promote the sequential incorporation of D-Tyrosine (D-Tyr) and L-2,4 Diaminobutyrate (L-Dab), thus forming a tetrahydropyrimidine ring, i.e., the precursor of the chromophore [43][59].
Subsequently, the peptide is modified by the action of PvdI, an enzyme composed of four modules catalyzing the addition of D-Ser, L-Arg, D-Ser, and L-N5-formyl-N5-hydroxyornithine (L-hfOrn) [60]. Other auxiliary enzymes associated with the biosynthetic pathway are PvdH, PvdA, and PvdF, responsible for the formation of specific amino acids composing the PVDs backbone, such as L-Dab and L-hfOrn. In detail, PvdH promotes the L-Dab synthesis starting from L-aspartate β-semialdehyde, while PvdA and PvdF allow L-ornithine hydroxylation and formylation yielding L-fOHOrn [61][62]. Interestingly, Gasser and colleagues [63] showed, using the FRET–FLIM (Förster resonance energy transfer measured by fluorescence lifetime microscopy) technique, that PvdA interacts physically with PvdJ, PvdI, PvdL, and PvD, suggesting the presence of a strongly organized multienzymatic complex coordinating the PVD biosynthesis. Similarly, Philem and collaborators [64] demonstrated the fundamental role played by PvdF through a mutagenesis experiment, as the mutants carrying the inactivated enzyme had a reduced production of PVDI. Therefore, the authors suggest a possible switch to regulate PVD production for biotechnological purposes.
As just mentioned, two other NRPSs directly involved in the elongation of the peptide are PvdJ and PvdD, catalyzing the addition of L-Lys, L-hfOrn, and two L-Thr residues [63][64][65]. The activity of two auxiliary enzymes, PvdG and MbtH, appears to be required for the intra-cytoplasmic maturation of the ferribactin. In detail, MbtH has been proven to enhance the adenylation activity by A domains from many NRPS enzymes [66], while the role of PvdG has not yet been fully elucidated. PvdG could be a trans-acting thioesterase for PvdL and/or PvdI [48]. In addition, in specific strains, different auxiliary enzymes can catalyze tailoring reactions during ferribactin biogenesis. As an example, during the biosynthesis of type II PVD, PvdYII catalyzes the acetylation of N-hydroxy-ornithine, which is supposed to be the preliminary step for the formation of the terminal N-hydroxy-cyclo-ornithine residue, and the subsequent peptide release from the NRPS machinery [55][67]. The acylated ferribactin is then transported to the periplasm through the activity of PvdE, a specific ABC transporter homolog to MacB ABC transporter for cyclic peptides [68]. In the periplasm, PVDI is subjected to diacylation by Ntn-type hydrolase PvdQ [69].
To achieve a functional pyoverdine, two copper-dependent oxidoreductases, PvdP and PvdO, provide for the cyclization and the formation of the final PVDI chromophore by catalyzing the first and last oxidative step, respectively [70][71]. The correct functioning of PvdP requires the activity of two auxiliary enzymes. The activity of PvdP was, in fact, closely associated with the presence of PvdM, which is believed to be indispensable for the incorporation of copper within PvdP [72]. The action of the PvdP seems to be subordinated even to the activity of one periplasmic membrane-associated oxidoreductase, CcmC, which transfers the electrons generated during oxidation to a periplasmatic redox-active compound [48]. However, further mutagenicity studies are required to better elucidate the role played by CcmC.
After the formation of the mature chromophore, the peptide undergoes a rearrangement of the side chains, especially at the level of the α-carboxy and α-amino groups of the L-Glu present in position 1, which can be replaced, depending on the strains investigated, by succinamide, succinate, or α-ketoglutarate, and, less frequently, malamide and malic acid, or even traces of intramolecular cyclized succinic acid. Little is known about why these modifications are made, but it is predicted that they can lead to an advantage according to the different environmental conditions surrounding the microorganism [55][73]. The first enzyme discovered as having an active role in the modification of side chains is PvdN, necessary for the addition of succinamide and succinic acid [73]. Similarly, the PtaA enzyme, periplasmic transaminase A, was associated with the presence of α-ketoglutarate on the PVDI side chain [74]. Although malamide and its derivative, malate, can probably be produced starting from succinamide, the biosynthesis of the two compounds remains currently unknown.
Finally, once the side chain replacements are completed, the mature PVDI is secreted from the periplasm into the external environment through efflux pumps. To date, only two ATP-dependent efflux pumps are known, PvdRT-OpmQ and MdtABC-OpmB. These mediate the passage of PVDI through the membrane [46][75]. However, it is certain that other transporters play a role in the transport of PVDs to the outside environment, because, following the creation of Pseudomonas strains with deletions for the two pumps, PVD secretion was not interrupted.
References
- Gessard, M. Sur La Fonction Fluorescigène Des Microbes. Ann. Inst. Pasteur Paris 1892, 6, 801–823.
- Meyer, J.M.; Abdallah, M. The Fluorescentpigment OfPseudomonas Fluorescens: Biosynthesis, Puri-Fication and Physicochemical Properties. J. Gen. Microbiol. 1978, 107, 319–328.
- Meyer, J.M.; Hornspreger, J. Role of Pyover-DinePfthe Iron Binding Fluorescent Pigment OfPseu-Domonas Fluorescensiron Transport. J. Gen. Microbiol. 1978, 107, 329–331.
- Mular, A.; Shanzer, A.; Kozłowski, H.; Hubmann, I.; Misslinger, M.; Krzywik, J.; Decristoforo, C.; Gumienna-Kontecka, E. Cyclic Analogs of Desferrioxamine e Siderophore for 68Ga Nuclear Imaging: Coordination Chemistry and Biological Activity in Staphylococcus Aureus. Inorg. Chem. 2021, 60, 17846–17857.
- Swayambhu, G.; Bruno, M.; Gulick, A.M.; Pfeifer, B.A. Siderophore Natural Products as Pharmaceutical Agents. Curr. Opin. Biotechnol. 2021, 69, 242–251.
- Andrews, S.C.; Robinson, A.K.; Rodríguez-Quiñones, F. Bacterial Iron Homeostasis. FEMS Microbiol. Rev. 2003, 27, 215–237.
- Manck, L.E.; Park, J.; Tully, B.J.; Poire, A.M.; Bundy, R.M.; Dupont, C.L.; Barbeau, K.A. Petrobactin, a Siderophore Produced by Alteromonas, Mediates Community Iron Acquisition in the Global Ocean. ISME J. 2022, 16, 358–369.
- GAUSE, G.F. Recent Studies on Albomycin, a New Antibiotic. Br. Med. J. 1955, 2, 1177–1179.
- Brickman, T.J.; Hansel, J.-G.; Miller, M.J.; Armstrong, S.K. Purification, Spectroscopic Analysis and Biological Activity of the Macrocyclic Dihydroxamate Siderophore Alcaligin Produced by Bordetella Pertussis and Bordetella Bronchiseptica. Biometals 1996, 9, 191–203.
- Takahashi, A.; Nakamura, H.; Kameyama, T.; Kurasawa, S.; Naganawa, H.; Okami, Y.; Takeuchi, T.; Umezawa, H.; Iitaka, Y. Bisucaberin, a New Siderophore, Sensitizing Tumor Cells to Macrophage-Mediated Cytolysis. II. Physico-Chemical Properties and Structure Determination. J. Antibiot. 1987, 40, 1671–1676.
- Zhang, J.; Qi, L.; Chen, G.; Yin, W.-B. Discovery and Genetic Identification of Amphiphilic Coprogen Siderophores from Trichoderm Hypoxylon. Appl. Microbiol. Biotechnol. 2021, 105, 2831–2839.
- Ijiri, M.; Fujiya, M.; Konishi, H.; Tanaka, H.; Ueno, N.; Kashima, S.; Moriichi, K.; Sasajima, J.; Ikuta, K.; Okumura, T. Ferrichrome Identified from Lactobacillus Casei ATCC334 Induces Apoptosis through Its Iron-Binding Site in Gastric Cancer Cells. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2017, 39, 1010428317711311.
- Mukherjee, P.K.; Hurley, J.F.; Taylor, J.T.; Puckhaber, L.; Lehner, S.; Druzhinina, I.; Schumacher, R.; Kenerley, C.M. Ferricrocin, the Intracellular Siderophore of Trichoderma Virens, Is Involved in Growth, Conidiation, Gliotoxin Biosynthesis and Induction of Systemic Resistance in Maize. Biochem. Biophys. Res. Commun. 2018, 505, 606–611.
- Roosenberg, J.M.; Miller, M.J. Total Synthesis of the Siderophore Danoxamine. J. Org. Chem. 2000, 65, 4833–4838.
- Codd, R.; Richardson-Sanchez, T.; Telfer, T.J.; Gotsbacher, M.P. Advances in the Chemical Biology of Desferrioxamine B. ACS Chem. Biol. 2018, 13, 11–25.
- Yamanaka, K.; Oikawa, H.; Ogawa, H.-O.; Hosono, K.; Shinmachi, F.; Takano, H.; Sakuda, S.; Beppu, T.; Ueda, K. Desferrioxamine E Produced by Streptomyces Griseus Stimulates Growth and Development of Streptomyces Tanashiensis. Microbiology 2005, 151, 2899–2905.
- Zhai, C.; Summer, D.; Rangger, C.; Haas, H.; Haubner, R.; Decristoforo, C.; Fusarinine, C. A Novel Siderophore-Based Bifunctional Chelator for Radiolabeling with Gallium-68. J. Label. Compd. Radiopharm. 2015, 58, 209–214.
- Sokol, P.A.; Darling, P.; Lewenza, S.; Corbett, C.R.; Kooi, C.D. Identification of a Siderophore Receptor Required for Ferric Ornibactin Uptake in Burkholderia Cepacia. Infect. Immun. 2000, 68, 6554–6560.
- Müller, G.; Isowa, Y.; Raymond, K.N. Stereospecificity of Siderophore-Mediated Iron Uptake in Rhodotorula Pilimanae as Probed by Enantiorhodotorulic Acid and Isomers of Chromic Rhodotorulate. J. Biol. Chem. 1985, 260, 13921–13926.
- Cornish, A.S.; Page, W.J. The Catecholate Siderophores of Azotobacter Vinelandii: Their Affinity for Iron and Role in Oxygen Stress Management. Microbiology 1998, 144 7, 1747–1754.
- Dertz, E.A.; Stintzi, A.; Raymond, K.N. Siderophore-Mediated Iron Transport in Bacillus Subtilis and Corynebacterium Glutamicum. JBIC J. Biol. Inorg. Chem. 2006, 11, 1087–1097.
- O’Brien, I.G.; Cox, G.B.; Gibson, F. Biologically Active Compounds Containing 2,3-Duhydroxybenzoic Acid and Serine Formed by Escherichia Coli. Biochim. Biophys. Acta-Gen. Subj. 1970, 201, 453–460.
- Wen, Y.; Wu, X.; Teng, Y.; Qian, C.; Zhan, Z.; Zhao, Y.; Li, O. Identification and Analysis of the Gene Cluster Involved in Biosynthesis of Paenibactin, a Catecholate Siderophore Produced by Paenibacillus Elgii B69. Environ. Microbiol. 2011, 13, 2726–2737.
- Cornish, A.S.; Page, W.J. Production of the Triacetecholate Siderophore Protochelin by Azotobacter Vinelandii. Biometals 1995, 8, 332–338.
- Bister, B.; Bischoff, D.; Nicholson, G.J.; Valdebenito, M.; Schneider, K.; Winkelmann, G.; Hantke, K.; Süssmuth, R.D. The Structure of Salmochelins: C-Glucosylated Enterobactins of Salmonella Enterica. BioMetals 2004, 17, 471–481.
- Griffiths, G.L.; Sigel, S.P.; Payne, S.M.; Neilands, J.B. Vibriobactin, a Siderophore from Vibrio Cholerae. J. Biol. Chem. 1984, 259, 383–385.
- Münzinger, M.; Budzikiewicz, H.; Expert, D.; Enard, C.; Meyer, J.M. Achromobactin, a New Citrate Siderophore of Erwinia Chrysanthemi. Z. Für Nat. C 2000, 55, 328–332.
- Smith, M.J.; Neilands, J.B. Rhizobactin, a Siderophore from Rhizobium Meliloti. J. Plant Nutr. 1984, 7, 449–458.
- Drechsel, H.; Tschierske, M.; Thieken, A.; Jung, G.; Zähner, H.; Winkelmann, G. The Carboxylate Type Siderophore Rhizoferrin and Its Analogs Produced by Directed Fermentation. J. Ind. Microbiol. 1995, 14, 105–112.
- Konetschny-Rapp, S.; Jung, G.; Meiwes, J.; Zähner, H. Staphyloferrin A: A Structurally New Siderophore from Staphylococci. Eur. J. Biochem. 1990, 191, 65–74.
- Montgomerie, J.Z.; Bindereif, A.; Neilands, J.B.; Kalmanson, G.M.; Guze, L.B. Association of Hydroxamate Siderophore (Aerobactin) with Escherichia Coli Isolated from Patients with Bacteremia. Infect. Immun. 1984, 46, 835–838.
- Seyedsayamdost, M.R.; Traxler, M.F.; Zheng, S.-L.; Kolter, R.; Clardy, J. Structure and Biosynthesis of Amychelin, an Unusual Mixed-Ligand Siderophore from Amycolatopsis Sp. AA4. J. Am. Chem. Soc. 2011, 133, 11434–11437.
- Knosp, O.; Tigerstrom, M.; Page, W.J. Siderophore-Mediated Uptake of Iron in Azotobacter Vinelandii. J. Bacteriol. 1984, 159, 341–347.
- Chen, Y.; Unger, M.; Ntai, I.; McClure, R.A.; Albright, J.C.; Thomson, R.J.; Kelleher, N.L. Gobichelin A and B: Mixed-Ligand Siderophores Discovered Using Proteomics. Medchemcomm 2013, 4, 233–238.
- De Voss, J.J.; Rutter, K.; Schroeder, B.G.; Su, H.; Zhu, Y.; Barry, C.E., 3rd. The Salicylate-Derived Mycobactin Siderophores of Mycobacterium Tuberculosis Are Essential for Growth in Macrophages. Proc. Natl. Acad. Sci. USA 2000, 97, 1252–1257.
- Sonnenschein, E.C.; Stierhof, M.; Goralczyk, S.; Vabre, F.M.; Pellissier, L.; Hanssen, K.Ø.; de la Cruz, M.; Díaz, C.; de Witte, P.; Copmans, D.; et al. Pseudochelin A, a Siderophore of Pseudoalteromonas Piscicida S2040. Tetrahedron 2017, 73, 2633–2637.
- Cox, C.D.; Adams, P. Siderophore Activity of Pyoverdin for Pseudomonas aeruginosa. Infect. Immun. 1985, 48, 130–138.
- Dhungana, S.; Michalczyk, R.; Boukhalfa, H.; Lack, J.G.; Koppisch, A.T.; Fairlee, J.M.; Johnson, M.T.; Ruggiero, C.E.; John, S.G.; Cox, M.M.; et al. Purification and Characterization of Rhodobactin: A Mixed Ligand Siderophore from Rhodococcus Rhodochrous Strain OFS. BioMetals 2007, 20, 853–867.
- Perry, R.D.; Balbo, P.B.; Jones, H.A.; Fetherston, J.D.; Demoll, E. Yersiniabactin from Yersinia Pestis: Biochemical Characterization of the Siderophore and Its Role in Iron Transport and Regulation. Microbiology 1999, 145, 1181–1190.
- Sah, S.; Singh, R. Siderophore: Structural And Functional Characterisation—A Comprehensive Review. Agriculture 2015, 61, 97–114.
- Gulick, A.M. Nonribosomal Peptide Synthetase Biosynthetic Clusters of ESKAPE Pathogens. Nat. Prod. Rep. 2017, 34, 981–1009.
- Sayari, M.; van der Nest, M.A.; Steenkamp, E.T.; Soal, N.C.; Wilken, P.M.; Wingfield, B.D. Distribution and Evolution of Nonribosomal Peptide Synthetase Gene Clusters in the Ceratocystidaceae. Genes 2019, 10, 328.
- Schalk, I.J.; Guillon, L. Pyoverdine Biosynthesis and Secretion in Pseudomonas aeruginosa: Implications for Metal Homeostasis. Environ. Microbiol. 2013, 15, 1661–1673.
- D’Onofrio, A.; Crawford, J.M.; Stewart, E.J.; Witt, K.; Gavrish, E.; Epstein, S.; Clardy, J.; Lewis, K. Siderophores from Neighboring Organisms Promote the Growth of Uncultured Bacteria. Chem. Biol. 2010, 17, 254–264.
- Cordero, O.X.; Ventouras, L.-A.; DeLong, E.F.; Polz, M.F. Public Good Dynamics Drive Evolution of Iron Acquisition Strategies in Natural Bacterioplankton Populations. Proc. Natl. Acad. Sci. USA 2012, 109, 20059–20064.
- Bonneau, A.; Roche, B.; Schalk, I.J. Iron Acquisition in Pseudomonas aeruginosa by the Siderophore Pyoverdine: An Intricate Interacting Network Including Periplasmic and Membrane Proteins. Sci. Rep. 2020, 10, 120.
- Budzikiewicz, H.; Schäfer, M.; Fernández, D.U.; Matthijs, S.; Cornelis, P. Characterization of the Chromophores of Pyoverdins and Related Siderophores by Electrospray Tandem Mass Spectrometry. BioMetals 2007, 20, 135–144.
- Ringel, M.T.; Brüser, T. The Biosynthesis of Pyoverdines. Microb. Cell 2018, 5, 424–437.
- Albrecht-Gary, A.M.; Blanc, S.; Rochel, N.; Ocaktan, A.Z.; Abdallah, M.A. Bacterial Iron Transport: Coordination Properties of Pyoverdin PaA, a Peptidic Siderophore of Pseudomonas aeruginosa. Inorg. Chem. 1994, 33, 6391–6402.
- Cézard, C.; Farvacques, N.; Sonnet, P. Chemistry and Biology of Pyoverdines, Pseudomonas Primary Siderophores. Curr. Med. Chem. 2015, 22, 165–186.
- Meyer, J.M.; Neely, A.; Stintzi, A.; Georges, C.; Holder, I.A. Pyoverdin Is Essential for Virulence of Pseudomonas aeruginosa. Infect. Immun. 1996, 64, 518–523.
- Visca, P.; Imperi, F.; Lamont, I.L. Pyoverdine Siderophores: From Biogenesis to Biosignificance. Trends Microbiol. 2007, 15, 22–30.
- Gasser, V.; Guillon, L.; Cunrath, O.; Schalk, I.J. Cellular Organization of Siderophore Biosynthesis in Pseudomonas aeruginosa: Evidence for Siderosomes. J. Inorg. Biochem. 2015, 148, 27–34.
- Ronnebaum, T.A.; Lamb, A.L. Nonribosomal Peptides for Iron Acquisition: Pyochelin Biosynthesis as a Case Study. Curr. Opin. Struct. Biol. 2018, 53, 1–11.
- Schalk, I.J.; Rigouin, C.; Godet, J. An Overview of Siderophore Biosynthesis among Fluorescent Pseudomonads and New Insights into Their Complex Cellular Organization. Environ. Microbiol. 2020, 22, 1447–1466.
- Gross, H.; Loper, J.E. Genomics of Secondary Metabolite Production by Pseudomonas Spp. Nat. Prod. Rep. 2009, 26, 1408–1446.
- Reimer, J.M.; Haque, A.S.; Tarry, M.J.; Schmeing, T.M. Piecing Together Nonribosomal Peptide Synthesis. Curr. Opin. Struct. Biol. 2018, 49, 104–113.
- Hannauer, M.; Schäfer, M.; Hoegy, F.; Gizzi, P.; Wehrung, P.; Mislin, G.L.A.; Budzikiewicz, H.; Schalk, I.J. Biosynthesis of the Pyoverdine Siderophore of Pseudomonas aeruginosa Involves Precursors with a Myristic or a Myristoleic Acid Chain. FEBS Lett. 2012, 586, 96–101.
- Gasser, V.; Malrieu, M.; Forster, A.; Mély, Y.; Schalk, I.; Godet, J. Supra-Molecular Organization of the Pyoverdine Bio-Synthetic Pathway in Pseudomonas aeruginosa. bioRxiv 2018.
- Durán, O.; Ramos, C.; Chen, O.; Castillo, J.; de Mayorga, B.; de Chial, M. Pyoverdine as an Important Virulence Factor in Pseudomonas aeruginosa Antibiotic Resistance. In The Global Antimicrobial Resistance Epidemic-Innovative Approaches and Cutting-Edge Solutions; Téllez, D.G., Ed.; IntechOpen: Rijeka, Croatia, 2022.
- Olucha, J.; Meneely, K.M.; Chilton, A.S.; Lamb, A.L. Two Structures of an N-Hydroxylating Flavoprotein Monooxygenase: Ornithine Hydroxylase from Pseudomonas aeruginosa. J. Biol. Chem. 2011, 286, 31789–31798.
- Kenjić, N.; Hoag, M.R.; Moraski, G.C.; Caperelli, C.A.; Moran, G.R.; Lamb, A.L. PvdF of Pyoverdin Biosynthesis Is a Structurally Unique N(10)-Formyltetrahydrofolate-Dependent Formyltransferase. Arch. Biochem. Biophys. 2019, 664, 40–50.
- Gasser, V.; Malrieu, M.; Forster, A.; Mély, Y.; Schalk, I.J.; Godet, J. In Cellulo FRET-FLIM and Single Molecule Tracking Reveal the Supra-Molecular Organization of the Pyoverdine Bio-Synthetic Enzymes in Pseudomonas aeruginosa. Q. Rev. Biophys. 2020, 53, e1.
- Philem, P.; Kleffmann, T.; Gai, S.; Hawkins, B.C.; Wilbanks, S.M.; Lamont, I.L. Identification of Active Site Residues of the Siderophore Synthesis Enzyme PvdF and Evidence for Interaction of PvdF with a Substrate-Providing Enzyme. Int. J. Mol. Sci. 2021, 22, 2211.
- Winn, M.; Fyans, J.K.; Zhuo, Y.; Micklefield, J. Recent Advances in Engineering Nonribosomal Peptide Assembly Lines. Nat. Prod. Rep. 2016, 33, 317–347.
- Baltz, R.H. Function of MbtH Homologs in Nonribosomal Peptide Biosynthesis and Applications in Secondary Metabolite Discovery. J. Ind. Microbiol. Biotechnol. 2011, 38, 1747–1760.
- Lamont, I.L.; Martin, L.W.; Sims, T.; Scott, A.; Wallace, M. Characterization of a Gene Encoding an Acetylase Required for Pyoverdine Synthesis in Pseudomonas aeruginosa. J. Bacteriol. 2006, 188, 3149–3152.
- Greene, N.P.; Kaplan, E.; Crow, A.; Koronakis, V. Antibiotic Resistance Mediated by the MacB ABC Transporter Family: A Structural and Functional Perspective. Front. Microbiol. 2018, 9, 950.
- Drake, E.J.; Gulick, A.M. Structural Characterization and High-Throughput Screening of Inhibitors of PvdQ, an NTN Hydrolase Involved in Pyoverdine Synthesis. ACS Chem. Biol. 2011, 6, 1277–1286.
- Nadal-Jimenez, P.; Koch, G.; Reis, C.R.; Muntendam, R.; Raj, H.; Jeronimus-Stratingh, C.M.; Cool, R.H.; Quax, W.J. PvdP Is a Tyrosinase That Drives Maturation of the Pyoverdine Chromophore in Pseudomonas aeruginosa. J. Bacteriol. 2014, 196, 2681–2690.
- Ringel, M.T.; Dräger, G.; Brüser, T. PvdO Is Required for the Oxidation of Dihydropyoverdine as the Last Step of Fluorophore Formation in Pseudomonas Fluorescens. J. Biol. Chem. 2018, 293, 2330–2341.
- Sugue, M.-F. Towards Understanding the Mechanisms of the Periplasmic Pyoverdine Maturation. Ph.D. Thesis, Institutionelles Repositorium der Leibniz Universität Hannover, Hannover, Germany, 2022.
- Ringel, M.T.; Dräger, G.; Brüser, T. PvdN Enzyme Catalyzes a Periplasmic Pyoverdine Modification. J. Biol. Chem. 2016, 291, 23929–23938.
- Ringel, M.T.; Dräger, G.; Brüser, T. The Periplasmic Transaminase PtaA of Pseudomonas Fluorescens Converts the Glutamic Acid Residue at the Pyoverdine Fluorophore to α-Ketoglutaric Acid. J. Biol. Chem. 2017, 292, 18660–18671.
- Henríquez, T.; Stein, N.V.; Jung, H. PvdRT-OpmQ and MdtABC-OpmB Efflux Systems Are Involved in Pyoverdine Secretion in Pseudomonas Putida KT2440. Environ. Microbiol. Rep. 2019, 11, 98–106.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
2 times
(View History)
Update Date:
25 Nov 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No