 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yves Menezo | + 2344 word(s) | 2344 | 2020-12-16 07:34:48 | | | |
| 2 | Dean Liu | -559 word(s) | 1785 | 2020-12-17 03:01:42 | | | | |
| 3 | Kay Elder | -14 word(s) | 1771 | 2020-12-17 08:39:38 | | |
Video Upload Options
Methylation is a universal biochemical process which covalently adds methyl groups to a variety of molecular targets. It plays a critical role in two major global regulatory mechanisms, epigenetic modifications and imprinting, via methyl tagging on histones and DNA. During reproduction, the two genomes that unite to create a new individual are complementary but not equivalent. Methylation determines the complementary regulatory characteristics of male and female genomes. DNA methylation is executed by methyltransferases that transfer a methyl group from S-adenosylmethionine, the universal methyl donor, to cytosine residues of CG (also designated CpG). Histones are methylated mainly on lysine and arginine residues. The methylation processes regulate the main steps in reproductive physiology: gametogenesis, and early and late embryo development, and thus play a crucial role in the transmission of life.
1. Introduction
Methylation is a universal biochemical process which covalently adds methyl groups to a variety of molecular targets, including neurotransmitters, lipids, proteins, and DNA. DNA repair, protein function, and gene expression involve methylation; it plays a critical role in two major global regulatory mechanisms: epigenesis and imprinting, which are transcriptional silencing and regulation of imprinted genes During reproduction, the two genomes that unite to create a new individual are complementary but not equivalent. For this reason, complete parthenogenesis (oocytes cleaving to form embryos without fertilization by sperm) or androgenesis (embryos developing without genetic contribution from an oocyte) cannot result in development of viable progeny. Methylation processes that regulate epigenesis and imprinting determine the characteristics of the regulatory processes that differ between male and female genomes. Imprinting “tags” a chemical mark in order to silence or activate genes, which is usually parent-specific. It can be reversible in order to switch genes “on” or “off” in different organs or during certain time periods (e.g., during pregnancy). This differs from epigenesis, which was defined by Conrad Waddington in 1942 as “causal interactions between genes and their products which bring the phenotype into being”. An epigenetic trait is a “stable heritable phenotype resulting from changes in a chromosome without alterations in the DNA sequence” [1] Modifications due to imprinting are considered more robust than those resulting from epigenesis. In general, both generate a unique chromosomal chemical modification for each parent, leading to different expressions of genes located on these chromosomes. Whether an imprinted gene is expressed depends upon the sex of the parent genome, with patterns of expression that result from chemical modification of DNA and/or alteration of chromatin structure via posttranslational modification of histone residues.
Epigenetic gene regulation is heritable and is directed by several different parameters, including DNA methylation, chromatin remodeling as a result of posttranslational histones modifications, and RNA interference. DNA methylation is executed by methyltransferases that transfer a methyl group from S-adenosylmethionine, the universal methyl donor, to cytosine residues of CG (also designated CpG) dinucleotides. In mammals, DNA methyltransferase-1 (DNMT1) is the predominant enzyme responsible for maintaining methylation during the cell cycle after DNA replication. De novo methylation, for example acquisition of new imprint tags, linked to the environment during pregnancy is principally carried out by DNMT3A and B: it will be necessary for epigenetic resetting in germ cells.
2. A Reminder of the Methylation Process
S-adenosyl methionine (SAM) is the universal cofactor for methylation as the methyl group donor (Figure 1). Methionine adenosyltransferase (MAT) catalyzes SAM formation by linking methionine and ATP. Methylation is then carried out by DNA methyltransferase and histone methyltransferase enzymes. After the targets have been methylated, S-adenosyl homocysteine (SAH) is formed, and homocysteine (HCY) is then released. Hcy must be recycled: it is a toxic metabolite that inhibits the methylation process and can also inactivate some proteins via homocysteinilation, which leads to structural modifications. The one-carbon cycle is the major cellular pathway that recycles Hcy, together with methionine synthase associated with the folate cycle (see Figure 1). Methyl tetrahydrofolate (MTHF) is the active compound for folate cycle methionine synthase activity, and correct methylation depends upon a supply of MTHF. Some types of cells can also recycle Hcy via the cystine beta synthase (CBS) pathway, releasing cysteine. A third mechanism, available primarily in liver cells but only marginal in other cells, uses the betaine homocysteine pathway. Cysteine plays an important role in the redox homeostasis that is necessary for correct methylation, and errors in methylation are heavily linked to unbalanced oxidative stress. Abnormally low/inadequate concentrations of methionine and cystine may have an effect on epigenetic processes, and mechanisms for cellular transport and/or synthesis of methionine, cystine, glycine, and glutamic acid are crucial in supporting correct methylation. The availability of these compounds for direct (methionine) or indirect (via the synthesis of glutathione) participation in methylation as well as protection of the process are essential for correct establishment and maintenance of epigenetic and imprinting methyl tags.
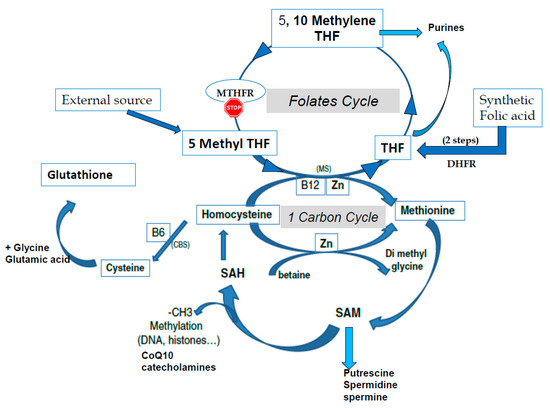
Figure 1. The one-carbon cycle (1-CC) and the folates cycle (MTHFR: methyltetrahydrofolate reductase, THF tetrahydrofolate). CBS: Cystathionine beta synthase, CoQ10: Coenzyme Q10, CH3: methyl group, DHFR: Dihydrofolate reductase, MS: methionine synthase, SAM S Adenosyl Methionine, SAH: S Adenosyl Homocysteine, Zn: Zinc.
3. The Beginning of Life: Fertilization and Immediately Postfertilization
During oogenesis, the oocyte accumulates epigenetic marks both on histones and on DNA, and this process continues until just before ovulation. At the time of fertilization, major intermediate metabolic changes are immediately initiated, which are necessary to support correct epigenetic/methylation status. All of the regulatory processes are strictly dependent upon maternal reserves of proteins and messenger RNAs (mRNAs) stored during oocyte growth; no transcription takes place in the newly fertilized egg until the zygote genome is activated (maternal to zygotic transition, MZT) at the 4- to 8-cell stages in human embryos. Interference RNAs (iRNAs) play an important role in regulating the translation of stored polyadenylated mRNAs.
Upregulation of the pentose phosphate pathway permits the formation of NADPH, essential for glutathione (GSH) synthesis: γ-L-Glutamyl-L-cysteinylglycine is a universal antioxidant molecule. GSH is necessary for sperm head swelling by opening the protamines that padlock the DNA and in order to protect the methylation process and to maintain redox homeostasis in order to prevent errors in methylation linked to unbalanced oxidative stress. As mentioned previously, abnormally low/inadequate concentrations of methionine and cystine may affect epigenetic processes [2]. Glutaredoxins are red-ox enzymes, classified as “light proteins”, that use glutathione as a cofactor. These proteins are thiol-disulfide oxidoreductases with a glutathione-binding site and one or two active cysteines in their active site. They can reduce methionine sulfone (oxidized methionine) to active methionine. Glutaredoxins are oxidized by oxidized substrates and are nonenzymatically reduced by reduced glutathione, which in turn is oxidized but can be regenerated by glutathione reductase (GRX). In terms of physio-biochemistry, glutaredoxins are present and highly expressed in early embryos: they actively protect redox homeostasis and thus have an impact on imprinting processes. Hypotaurine (Htau) is also found in the in vivo embryonic environment as an efficient antioxidant synthesized by oviductal cells and released into the tubal lumen[3][4].
Oocytes and early embryos have active mechanisms for cellular transport and/or synthesis of methionine, cystine, glycine, and glutamic acid. SAM is actively synthesized, and all of the enzymes involved in the methylation process are present, including the methionine synthase pathway that regenerates methionine from Hcy. The expression of CBS pathway enzymes is weak or absent, which means that homocysteine cannot be recycled to cystine, reinforcing the requirement for cystine and methionine. The betaine homocysteine pathway (BHMT) is only marginally represented. Human oocytes express high levels of folate receptor 1 and folate transporter1 (SLC19A1), indicating that these molecules play an important role during the first three to four days of development prior to the onset of genomic activation, MZT. Folates are central to a system that involves high molecular trafficking[5], and all of the enzymes involved in the folate and 1-carbon cycles are highly expressed in the oocyte. The embryo also finds important metabolites in its tubal fluid environment.
A major upheaval occurs during and immediately after the fertilization period: the DNA of the zygote genome is thought to be rapidly de-methylated immediately postfertilization, and human in vitro fertilized IVF embryos apparently follow this scheme. High sperm methylation and retained methylation of the paternal genome represent major markers of fertility[6][7][8]. The observation that isoforms affecting the one-carbon and folate cycles have a negative impact on fertility confirms the importance of a high methylation status in sperm. Paternal effects on early embryonic development is a feature that is well documented in bovine embryos. Suboptimal sperm methylation in terms of “quality and quantity” (CpG island regions restricted to retained histones) has a negative impact on human embryo blastocyst development[9] Approximately 10–15% of histones are retained, but these are not randomly located: they bind to the “active part/coding area” of DNA and so play an important role in embryonic development [6][9].
As mentioned earlier, global methylation in the sperm nucleus (DNA and histones) provides the key for correct spatial and biochemical conformation that will allow rapid access to the paternal genome after nuclear swelling in the sperm head[7][8]. This feature is mandatory for rapid S-phase activation and the major methylation/epigenetic modifications that will modify the male genome. Minor epigenetic alterations in sperm will immediately affect the early stages of preimplantation embryo developmental capacity. The paternal genome is rapidly demethylated initially, whereas the maternal genome is passively demethylated slowly during the subsequent cell cycle. The majority of the epigenetic methyl tags should be erased during de-methylation, but those that are retained and transmitted to the offspring probably transmit important epigenetic and imprint information. In human embryos, just prior to and during the second half of the pronuclear stage and before entering the first mitotic division, demethylated paternal DNA is immediately re-methylated, together with de novo H3-K9 tri-methylation[10]. A similar feature is observed in mouse, rabbit, and pig embryos. Oocytes have a significant endogenous pool of polyadenylated mRNAs coding for DNMT1 (DNA methyltransferase1), which is responsible for maintenance of methylation[3][5][11][12].
Global demethylation during preimplantation embryo development excludes imprinted genes: the parental imprints must be protected by DNMT1. The expression of DNMT3, responsible for de novo methylation, is weaker.
Two major issues must be highlighted: maternal age and the environment and the significant genetic impact of mutations in the one-carbon and folate cycles[13][14][15] The negative impact of abnormal methylation on bovine blastocyst formation has been documented[16]. Methylene tétrahydrofolate reductase (MTHFR) knockout decreases the number of blastocysts obtained, and those remaining have fewer cells in both the inner cell mass and the trophectoderm. Older females have a reduced abundance of DNA methyltransferases in MII oocytes, as the majority of important oocyte mRNAS are involved in regulation of homeostasis (redox, intermediate metabolism, etc.). Incorrect regulation of DNA methylation will further lead to inappropriate gene expression[17].
References
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783.
- Ménézo, Y.; Elder, K. Epigenetic remodeling of chromatin in human ART: Addressing deficiencies in culture media. J. Assist. Reprod. Genet. 2020, 37, 1781–1788.
- Guérin, P.; El Mouatassim, S.; Ménézo, Y. Oxidative stress and protection against reactive oxygen species in the pre-implantation embryo and its surroundings. Hum. Reprod. Update 2001, 7, 175–189.
- Guérin, P.; Ménézo, Y. Hypotaurine and taurine in gamete and embryo environments: De novo synthesis via the cysteine sulfinic acid pathway in oviduct cells. Zygote 1995, 3, 333–337.
- Ménézo, Y.; Lichtblau, I.; Elder, K. New insights into human pre-implantation metabolism in vivo and in vitro. J. Assist. Reprod. Genet. 2013, 30, 293–303.
- Denomme, M.M.; McCallie, B.R.; Parks, J.C.; Schoolcraft, W.B.; Katz-Jaffe, M.G. Alterations in the sperm histone-retained epigenome are associated with unexplained male factor infertility and poor blastocyst development in donor oocyte IVF cycles. Hum. Reprod. 2017, 32, 2443–2450.
- Ward, W.S. Function of sperm chromatin structural elements in fertilization and development. Mol. Hum. Reprod. 2010, 16, 30–36.
- Kutchy, N.A.; Menezes, E.S.B.; Ugur, M.R.; Ul Husna, A.; ElDebaky, H.; Evans, H.C.; Beaty, E.; Santos, F.C.; Tan, W.; Wills, R.W.; et al. Sperm cellular and nuclear dynamics associated with bull fertility. Anim. Reprod. Sci. 2019, 211, 106203.
- Ihara, M.; Meyer-Ficca, M.L.; Leu, N.A.; Rao, S.; Li, F.; Gregory, B.D.; Zalenskaya, I.A.; Schultz, R.M.; Meyer, R.G. Paternal poly (ADP-ribose) metabolism modulates retention of inheritable sperm histones and early embryonic gene expression. PLoS Genet. 2014, 10, e1004317.
- Park, J.S.; Jeong, Y.S.; Shin, S.T.; Lee, K.K.; Kang, Y.K. Dynamic DNA methylation reprogramming: Active demethylation and immediate re-methylation in the male pronucleus of bovine zygotes. Dev. Dyn. 2007, 236, 2523–2533.
- Menezo, Y.; Clement, P.; Dale, B. DNA Methylation Patterns in the Early Human Embryo and the Epigenetic/Imprinting Problems: A Plea for a More Careful Approach to Human Assisted Reproductive Technology (ART). Int. J. Mol. Sci. 2019, 20, 1342.
- Uysal, F.; Akkoyunlu, G.; Ozturk, S. Dynamic expression of DNA methyltransferases (DNMTs) in oocytes and early embryos. Biochimie 2015, 116, 103–113.
- Hara-Isono, K.; Matsubara, K.; Mikami, M.; Arima, T.; Ogata, T.; Fukami, M.; Kagami, M. Assisted reproductive technology represents a possible risk factor for development of epimutation-mediated imprinting disorders for mothers aged ≥30 years. Clin. Epigenet. 2020, 12, 111.
- Enciso, M.; Sarasa, J.; Xanthopoulou, L.; Bristow, S.; Bowles, M.F.; Fragouli, E.; Delhanty, J.; Wells, D. Polymorphisms in the MTHFR gene influence embryo viability and the incidence of aneuploidy. Hum. Genet. 2016, 135, 555–568.
- Servy, E.J.; Jacquesson-Fournols, L.; Cohen, M.; Menezo, Y. MTHFR isoform carriers. 5-MTHF (5-methyl tetrahydrofolate) vs. folic acid: A key to pregnancy outcome: A case series. J. Assist. Reprod. Genet. 2018, 35, 1431–1435.
- Ishitan, H.; Ikeda, S.; Egashira, K.; Sugimoto, M.; Kume, S.; Minami, N.; Ohta, T.J. Embryonic MTHFR contributes to blastocyst development. J. Assist. Reprod. Genet. 2020, 37, 1807–1814.
- Marshall, K.L.; Rivera, R.M. The effects of superovulation and reproductive aging on the epigenome of the oocyte and embryo. Mol. Reprod. Dev. 2018, 85, 90–105.

