Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | José Pérez de la Lastra | -- | 3080 | 2022-11-22 14:41:06 | | | |
| 2 | Amina Yu | + 4 word(s) | 3084 | 2022-11-23 01:39:35 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Andrés, C.M.C.; Lastra, J.M.P.D.L.; Juan, C.A.; Plou, F.J.; Pérez-Lebeña, E. Reactive Species on Amino Acids. Encyclopedia. Available online: https://encyclopedia.pub/entry/35859 (accessed on 21 July 2026).
Andrés CMC, Lastra JMPDL, Juan CA, Plou FJ, Pérez-Lebeña E. Reactive Species on Amino Acids. Encyclopedia. Available at: https://encyclopedia.pub/entry/35859. Accessed July 21, 2026.
Andrés, Celia María Curieses, José Manuel Pérez De La Lastra, Celia Andrés Juan, Francisco J. Plou, Eduardo Pérez-Lebeña. "Reactive Species on Amino Acids" Encyclopedia, https://encyclopedia.pub/entry/35859 (accessed July 21, 2026).
Andrés, C.M.C., Lastra, J.M.P.D.L., Juan, C.A., Plou, F.J., & Pérez-Lebeña, E. (2022, November 22). Reactive Species on Amino Acids. In Encyclopedia. https://encyclopedia.pub/entry/35859
Andrés, Celia María Curieses, et al. "Reactive Species on Amino Acids." Encyclopedia. Web. 22 November, 2022.
Copy Citation
Reactive oxygen species ROS can be triggered by exogenous sources (tobacco, pollution, xenobiotics, drugs, ionizing radiation and so on), but they can also be generated inside the cell by two different mechanisms: enzymatic and non-enzymatic; in both cases, they can have irreversible effects on animal and plant cells and tissues. The superoxide anion •O2− is unstable and cannot pass through membranes, but is rapidly converted to hydrogen peroxide H2O2 and it is membrane-permeable. In the Fenton reaction, H2O2 produces the hydroxyl radical •OH + −OH, which is highly reactive in the mitochondrial matrix. Elevated levels of ROS lead to increased mtDNA damage.
reactive species
ROS
RNS
RHS
amino acids
proteins
1. Reactive Stress on Amino Acids
In recent decades, there has been considerable interest in the idea that chronic oxidative/nitrosative stress plays a key role in the etiology of human disease [1]. These reactions, mediated by reactive oxygen species ROS and reactive nitrogen species RNS, are detrimental to cellular function, but do not present detectable disease-triggering symptoms [2]. Enhanced protection systems against oxygen and nitrogen radicals are thought to play a key role in primate evolution, resulting in increased longevity and lower rates of age-related diseases [3].
Peroxynitrite ONOO− is an oxidant and nitrating agent that can penetrate biological membranes [4]. Peroxynitrite reacts with proteins through three possible pathways: (i) peroxynitrite reacts directly with cysteine, methionine, and tryptophan; (ii) peroxynitrite reacts rapidly with transition metal centers and selenium-containing amino acids; and (iii) free radicals formed during homolysis of peroxynitrite, such as hydroxyl and nitrogen dioxide radicals, and the carbonate radical •CO3− formed in the presence of carbon dioxide, also react with proteins. •CO3− is negatively charged in all physiological environments, including those of acidic pH such as the phagolysosomes of phagocytic cells and ischemic tissues. Although less oxidizing than the hydroxyl radical (E0 = 2.3 V, pH 7.0), the carbonate radical (E0 = 1.78 V, pH 7.0) is a very strong one-electron oxidant that acts by both electron transfer and hydrogen abstraction mechanisms to produce radicals from the oxidized targets. The inability of the carbonate radical to produce stable adducts makes it difficult to prove its production under physiological conditions. In contrast, other radicals, such as the hydroxyl radical and nitrogen dioxide, produce stable adducts with exogenous targets and biomolecules.
As a result, the changes caused by these radicals are more complex than those caused by hydroxyl radicals alone, since they lead to oxidation and nitration.
In vivo, protein nitration at the tyrosine residue is considered a biomarker for nitrosative stress. The oxidative stress biomarker, 3-nitrotyrosine, is useful for identifying major neurodegenerative diseases [5]. In this reaction, nitrotyrosine units are generated in peptides and proteins by a radical reaction. The nitration of tyrosine begins with the formation of the tyrosyl radical, which is formed when tyrosine is oxidized by the hydroxyl or carbonate radical. Following the mechanism of nitration of tyrosine already documented in the literature, the chemical mechanism of hydroxylation starts with the formation of the phenoxyl radical in the aromatic ring, just as in the nitration of tyrosine.
Although the vast majority of research on RNS-derived protein modifications has long focused on the nitration of Tyr residues, there is increasing evidence that nitrated or hydroxylated tryptophan may also play an important role in controlling cellular processes. Tryptophan bound to proteins is susceptible to modification by nitrating agents, and it is important to determine the nature of these modifications. This is because the nitration and oxidation products can take different forms. Yamakura et al. (2005) found that ONOO induced Trp modifications in human Cu, Zn-SOD, including 5- and 6-NO2-Trp, and kynurenine, oxindole-3-alanine, and dihydroxy-tryptophan [6]. The 2007 study by Ishii et al. showed that ONOO− treatment leads to the formation of 2-, 4-, and 6-NO2-Trp, with 6-NO2-Trp being the most abundant product [7].
Protein S-nitrosylation SNO is a covalent modification of cysteine residues involving •NO, peroxynitrite and its derivatives. Proteins can undergo reversible post-translational modifications for which SNO is required. Accurate prediction of SNO sites is a topic that is currently receiving much attention, because it is very important for elucidating biological changes. Several studies have shown that S-nitrosylation controls both physiological and pathological processes, including immunological response and cellular senescence [8]. Alzheimer’s disease and breast cancer are two other diseases that may be due to the abnormal S-nitrosylation of proteins [9][10].
Methionine is one of the most easily oxidized amino acids with cysteine, tyrosine, and tryptophan. Peroxynitrous acid and peroxynitrite combine with methionine to form sulfoxides, and methionine catalyses the isomerization of peroxynitrite to nitrate. The pH and methionine concentration have no effect on the distribution of nitrite, nitrate, and methionine sulfoxide, the only detectable products [11]. This is a straight bimolecular process.
There is a growing interest in the post-translational modification of proteins by methionine sulfoxide [12]. Oxidation of methionine to sulfoxide can lead to significant structural and/or functional changes in protein activity, which can be up- or down-regulated. Methionine sulfoxide has been shown to serve as a conformational transition between -helical and -sheet forms in model peptides [12], a process similar to that observed in neurodegenerative diseases.
HOCl interacts rapidly with amines and, to a lesser extent, with amides, producing chlorinated derivatives such as chloramines (R-N(R’)-Cl) and chloramides (R-C(O)-N(R’)-Cl). These compounds can be further oxidized to produce dichlorinated species (R-N-Cl2). The α-amino group of amino acids, the N-terminus of peptides and proteins as well as nucleophilic sites on the side of protein chains can be chlorinated (e.g., lysine, arginine). Depending on the ratio of chlorine to amino acid, chlorination of amino acids leads to the formation of organic mono- or di-chloramines. Aldehydes, nitriles, and N-chlorodimines are all likely by-products of the degradation of organic chloramines, and all three pose a risk to human health. Chloramine production interferes with protein folding and causes protein aggregation [13].
2. Amino Acid Nitration and Hydroxylation
2.1. Tyrosine Nitration and Hydroxylation
Figure 1 shows the mechanism of tyrosine nitration described in the literature. Because of its short half-life of 10−2 s, peroxynitrite cannot react directly with tyrosine residues. Instead, it decomposes into oxidizing and nitrating species, including the radicals—•OH and•NO2. The radical •OH removes hydrogen from the phenol group of tyrosine. This produces the tyrosyl radical, which reacts with the •NO2 to form 3-nitrotyrosine 3- NTyr [14].
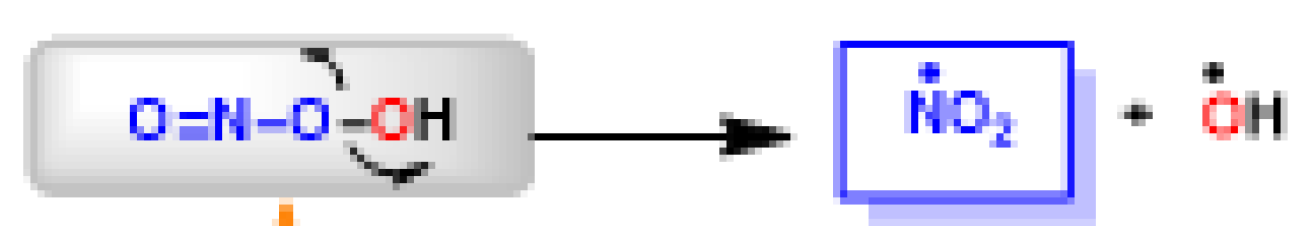
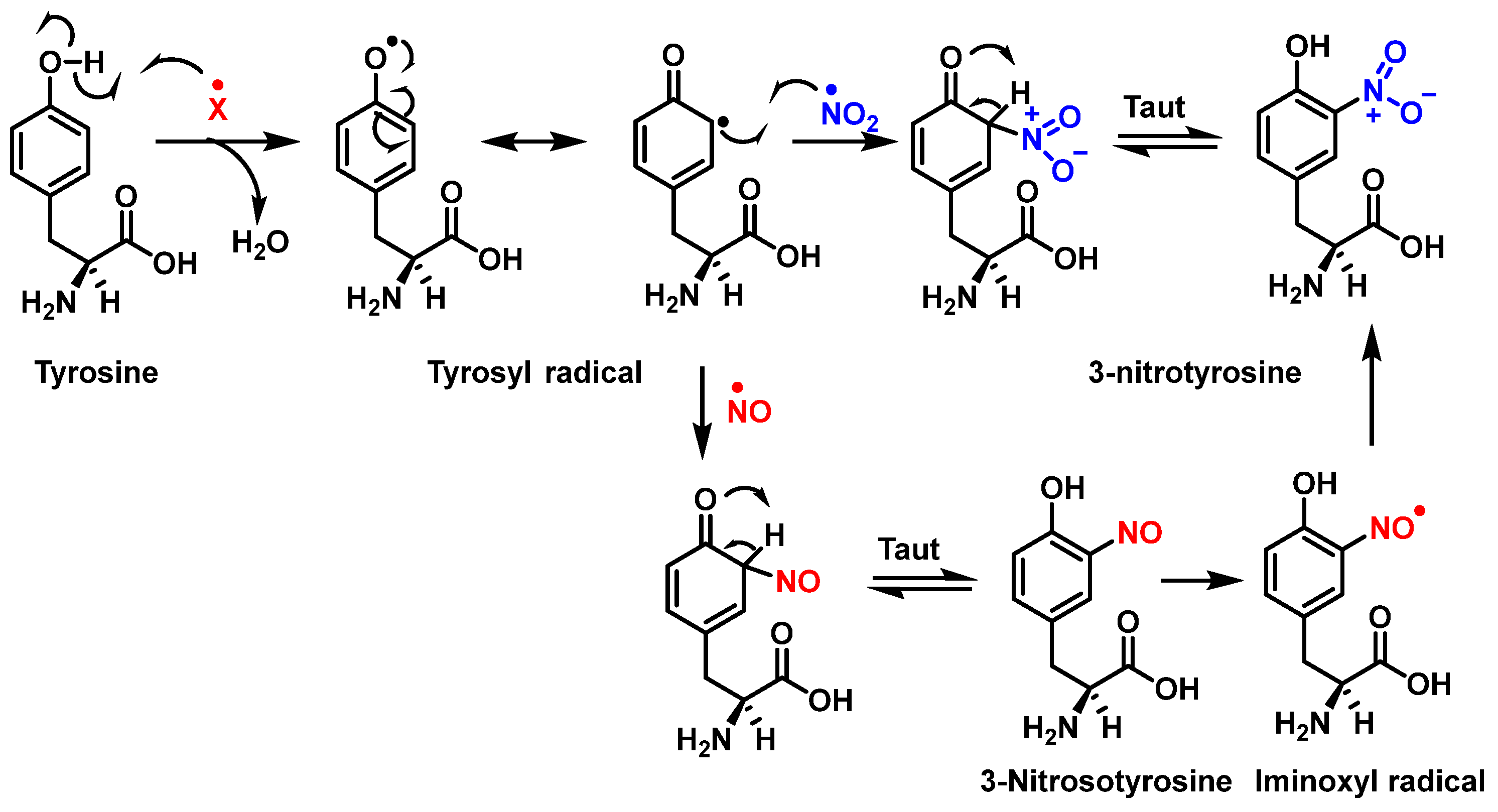
Figure 1. Mechanism of tyrosine nitration with either •NO2 (in blue) or •NO (in red) leading to the formation of 3-nitrotyrosine.
The tyrosyl radical formed during the oxidation of tyrosine by various oxidizing agents reacts with •NO2 to form 3-NTyr. It is also possible to nitrate tyrosine by an alternative route in which tyrosyl radicals react with •NO to form 3-nitrosotyrosine. Using an iminoxyl radical as an intermediate, oxo-metal complexes can further oxidize this product to form NO2-Tyr.
Hydroxylation of tyrosine follows a similar mechanism, in which •NO2 is replaced by •OH, Figure 2. In the mechanism, the •OH can be added to the tyrosine phenyl ring, producing a hydroxytyrosyl radical, stable by odd-electron delocalization, to generate 3-hydroxytyrosine.
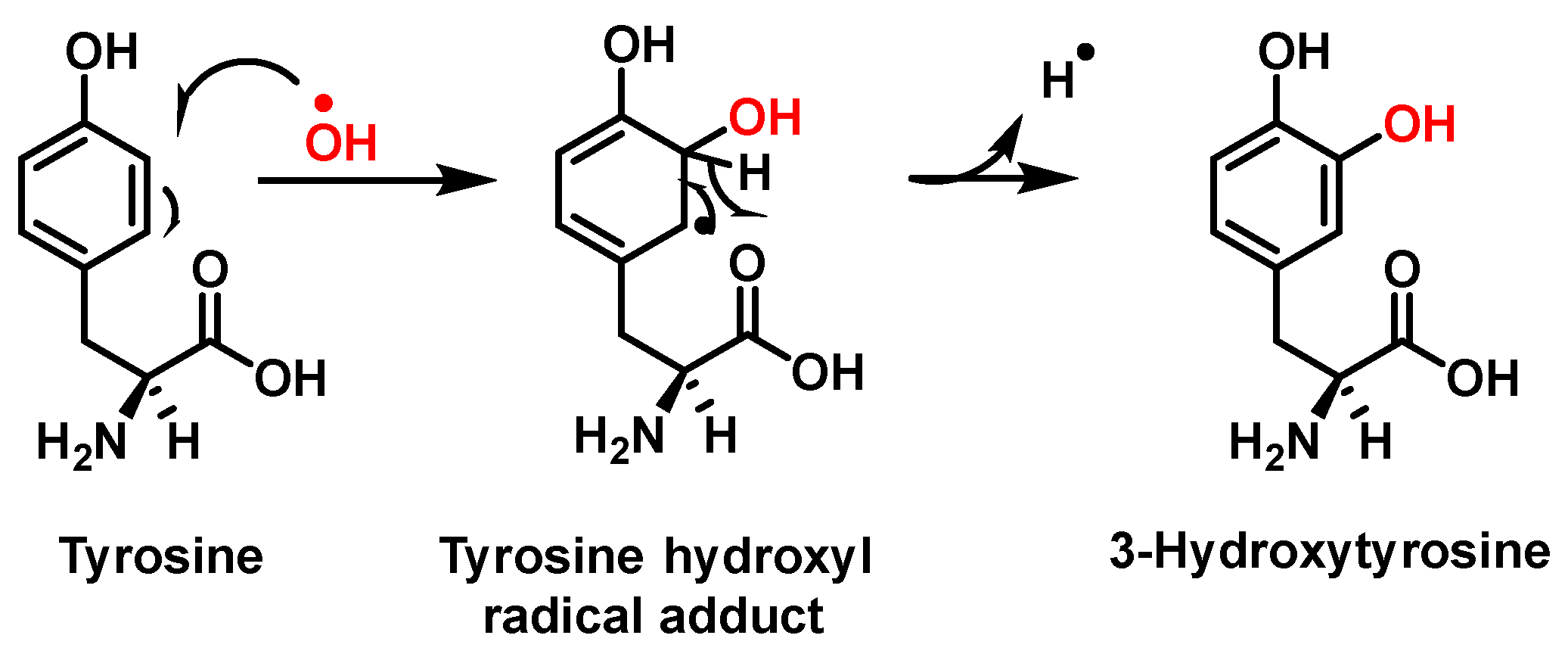
Figure 2. Mechanism of tyrosine hydroxylation with •OH leading to the formation of hydroxytyrosine.
The energy values of amino acids and its derivatives have been analyzed using the zwitterion structure, as this form can be found in the cell cytosol in equilibrium with the non- zwitterion structure, as its pH is usually maintained between 7.0 and 7.4 [15].
2.2. Tryptophan Nitration and Hydroxylation
Tryptophan nitration is less frequent than tyrosine nitration. In homogenized rat brain media, treated with 1 mM peroxynitrite, 244 peptides with nitrated tyrosine were identified, compared to only 2 peptides with nitrotryptophan [16]. The numbering of the carbon atoms in the aromatic ring of the tryptophan molecule is shown in Figure 3.
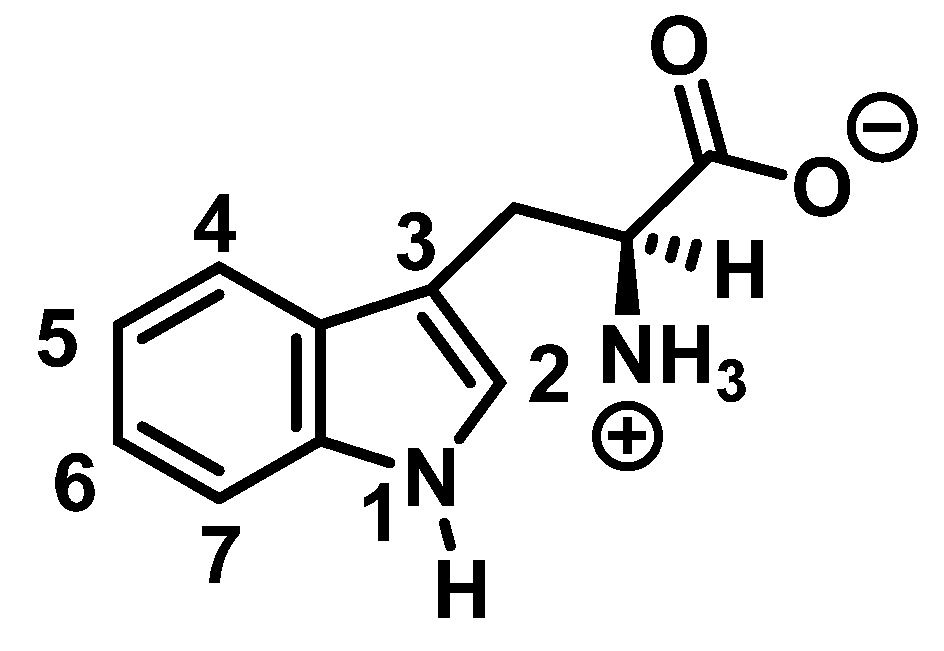
Figure 3. Numbering of the carbon atoms in the aromatic ring of the tryptophan molecule.
Although the mechanism of the reaction between ONOO and Trp is less clear, it probably follows a similar pattern to that described for the synthesis of 3-NTyr. Tryptophanyl radicals were observed after the addition of ONOO−, suggesting that ONOO− also modifies Trp via a radical intermediate. Unlike Tyr, whose indole can only be modified at a single carbon atom in the benzene ring, the indole of Trp has multiple reactive sites, including carbons 2-, 4-, 5-, 6- and 7 as well as nitrogen. Therefore, 1-, 2-, 3-, 4-, 5-, 6-, and 7-nitrotryptophan are all possible outcomes of the ONOO− reaction with Trp. In addition, the formation of a variety of oxidation products occurs, possibly due to a tryptophanyl radical at one of these sites.
In addition, Trp has been shown to nitrate in response to ONOO− treatment. This occurs when ONOO− combines with OH to form an ONOO radical, and subsequently decomposes to NO, which reacts with Trp. Trp, unlike Tyr residues, can react directly with ONOO− to produce other oxidation products. It is possible to nitrate tryptophan by first removing a proton in an aromatic electrophilic substitution, as shown in Figure 4, and then adding a nitronium ion-like species (+NO2) to the indole ring.
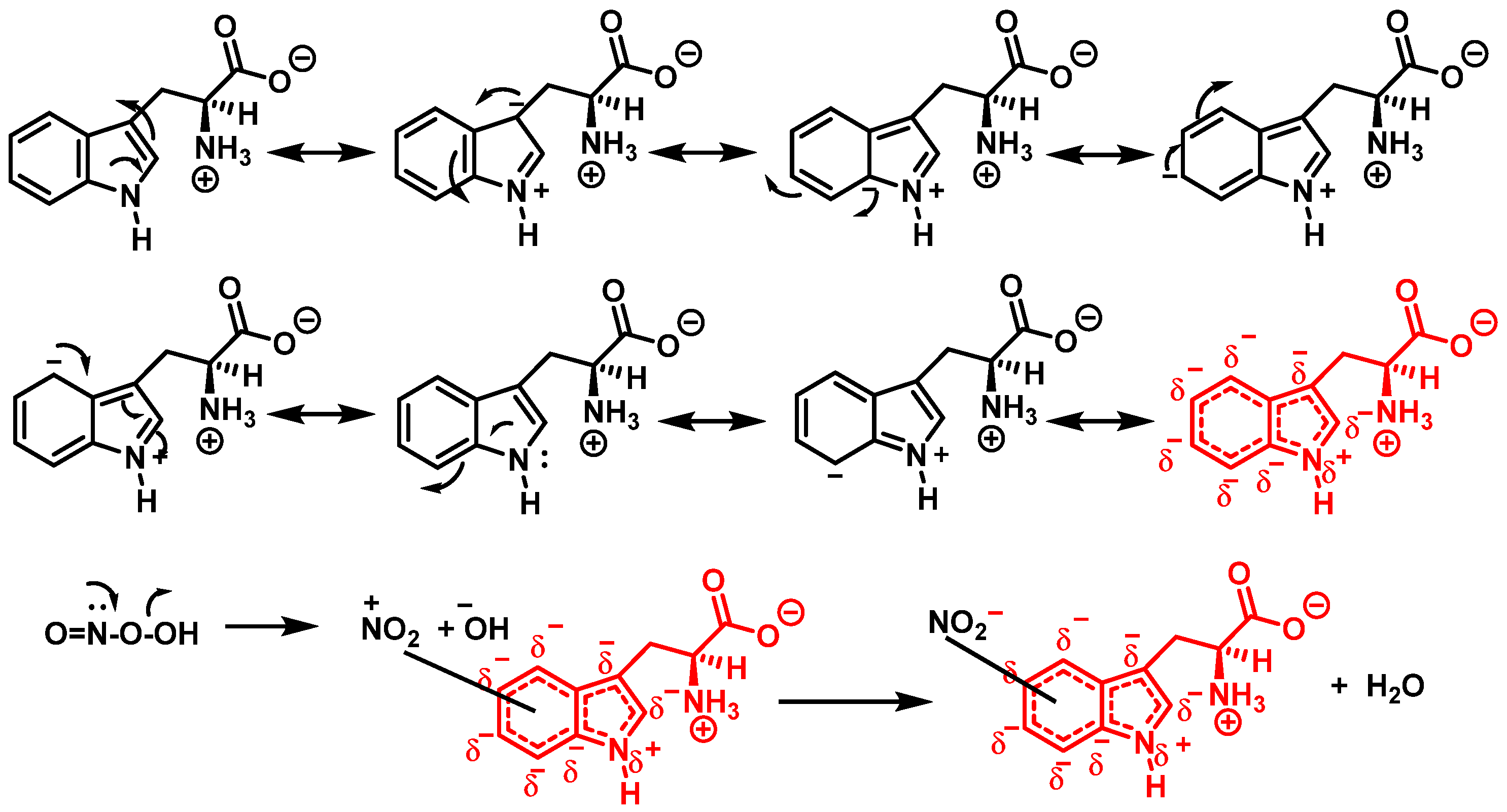
Figure 4. Mechanism for tryptophan nitration by addition of a nitronium ion-like species (+NO2) to the indole ring and removal of a proton in an aromatic electrophilic substitution.
Alternatively, nitration can be accomplished by removing the hydrogen atom from the nitrogen to produce a stabilized tryptophanyl radical capable of delocalizing the odd electron throughout the six-membered ring, followed by the addition of the nitrogen dioxide radical, either in steps or simultaneously, as shown in Figure 5 In this way, the one-electron oxidation of tryptophan is thermodynamically possible because the reduction potential E°′ (ONOO−,2H+/•NO2, H2O), +1.4 V [17], is higher than E°′ (tryptophanyl radical, H+/tryptophan), +1.0 V [18].
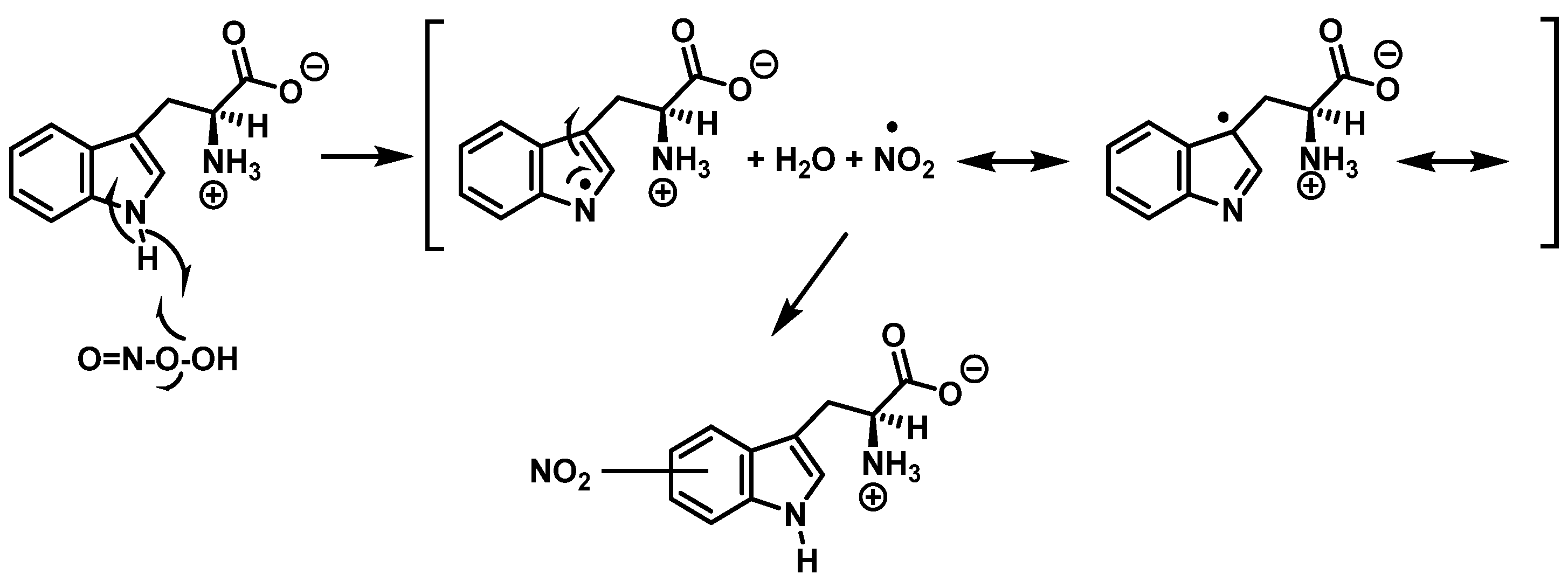
Figure 5. Alternative mechanism for tryptophan nitration via abstraction of the hydrogen atom from the nitrogen to give stabilized tryptophanyl radical, which can delocalize the odd electron throughout the ring, followed by addition of the •NO2 radical.
In summary, 6-nitrotryptophan is the major by-product of the reaction between peroxynitrite and tryptophan, followed by other nitrated and oxidized isomers such as N-formylkinurenine, and the more labile 1-nitrosotryptophan.
The peroxynitrite ion is stable, but its protonated form (ONOOH, pKa = 6.5 to 6.8) decomposes rapidly through homolysis to a variety of RNS. Its decomposition can form approximately 28% free •NO2 and •OH radicals [19], Figure 6.
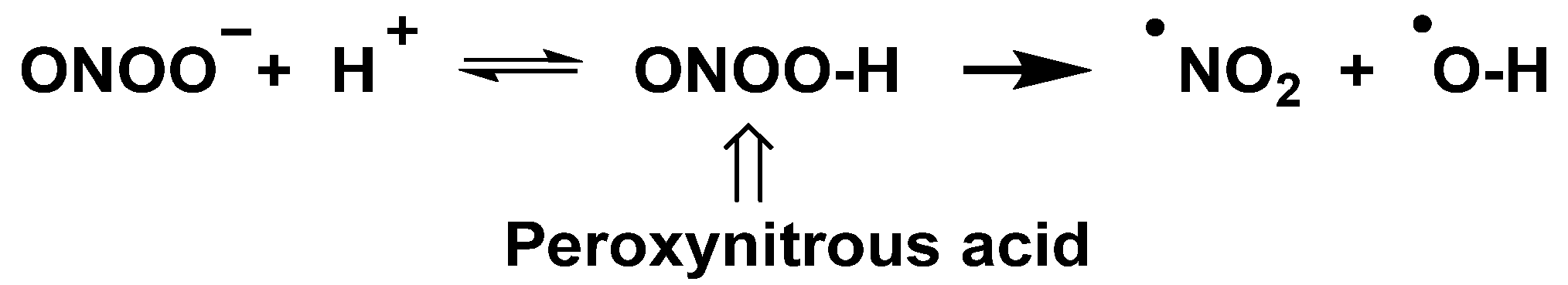
Figure 6. Decomposition of peroxynitrite to •NO2 and •OH.
Tryptophan can be converted to 2-, 4-, 5-, 6- or 7-hydroxyderivatives, and to N-formylkinurenine and kinurenine, Figure 7.
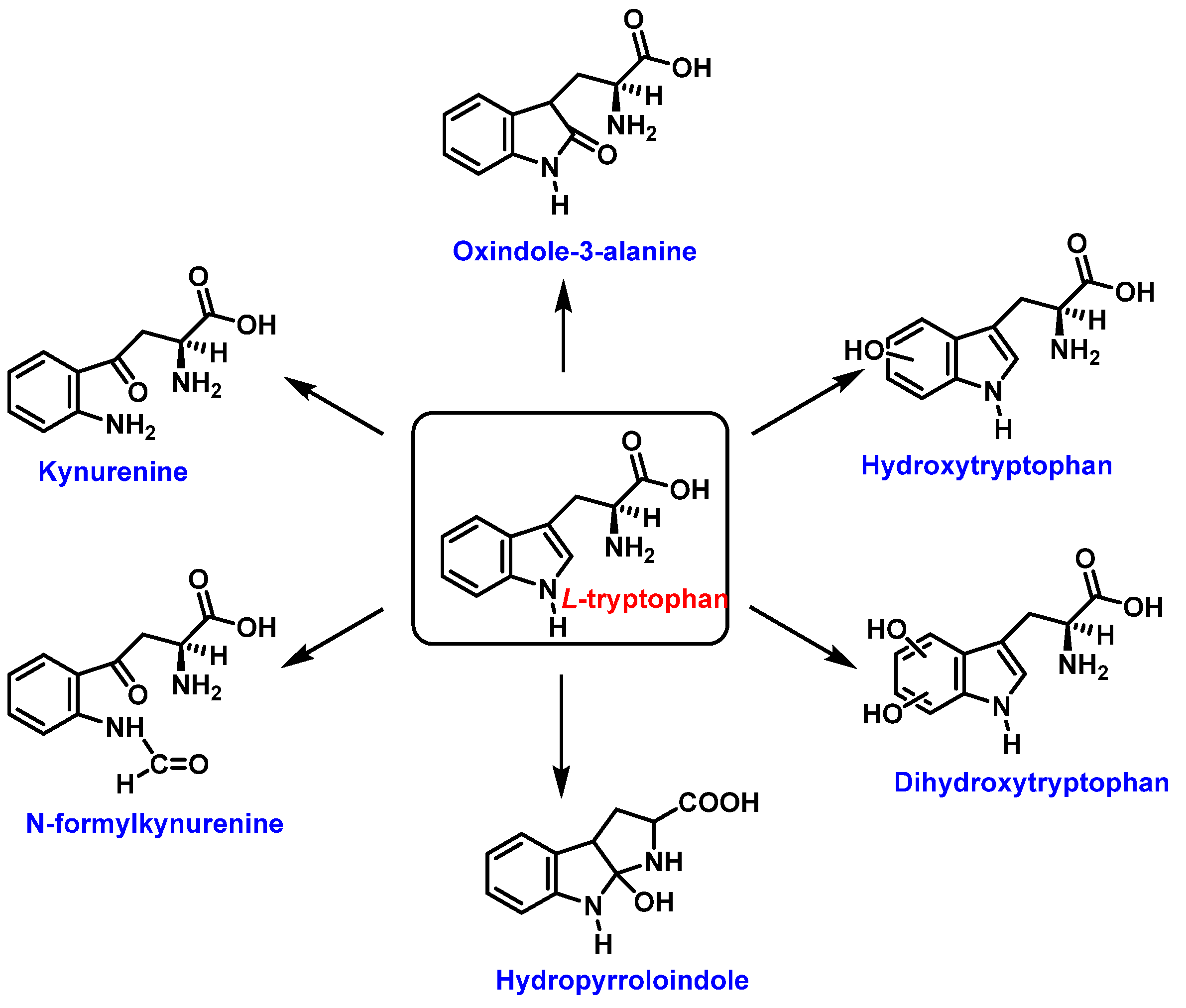
Figure 7. Oxidation products of L-tryptophan.
Addition of the •OH on the benzene ring and hydroxy isomers formation is represented in Figure 8.
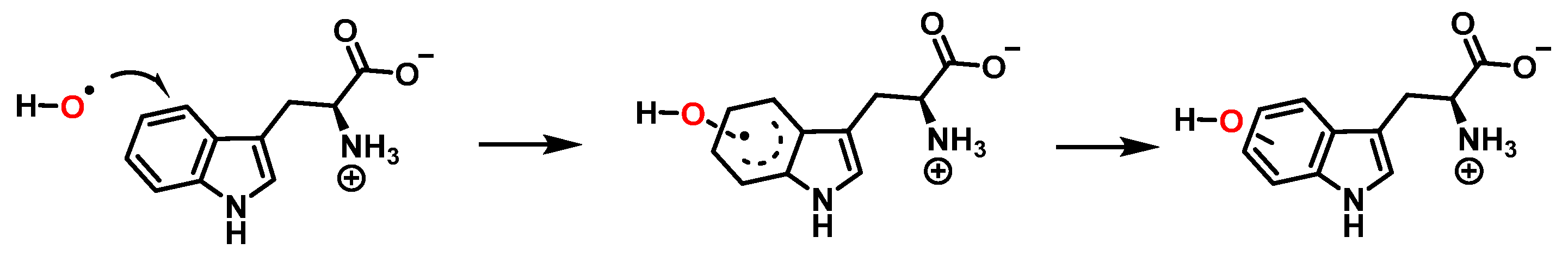
Figure 8. Formation of the hydroxy isomers.
For the formation of oxindole-3-alanine, there is the addition of the •OH to C-2 and formation of the radical at C-3, stabilized by being delocalized with the double bonds of the benzene ring. The last step is the formation of the enol in tautomeric equilibrium with the ketone form [20], Figure 9.
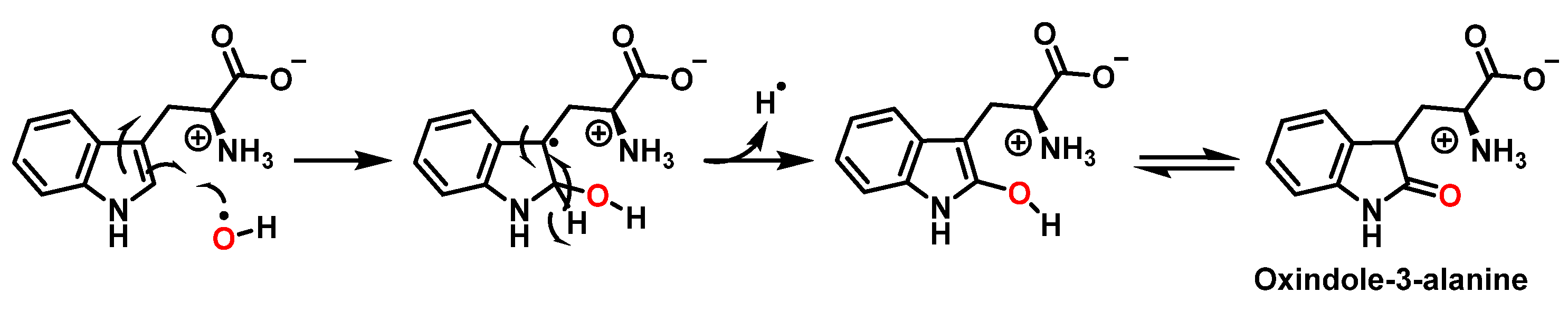
Figure 9. Oxindole-3-alanine formation mechanism.
The formation of hydropyrroloindole from oxindole-3-alanine could be explained by the nucleophilic addition of NH2 to the carbonyl carbon, generating an unstable NO-acetal, Figure 10. Kato et al. (1997) found that equimolar concentrations of ONOO− and tert-butoxycarbonyl-tryptophan (Boc-Trp) produced oxindole-3-alanine (Hydropyrroloindole and N-formylkinurenine) exclusively, without identifying nitrMetation products [20].
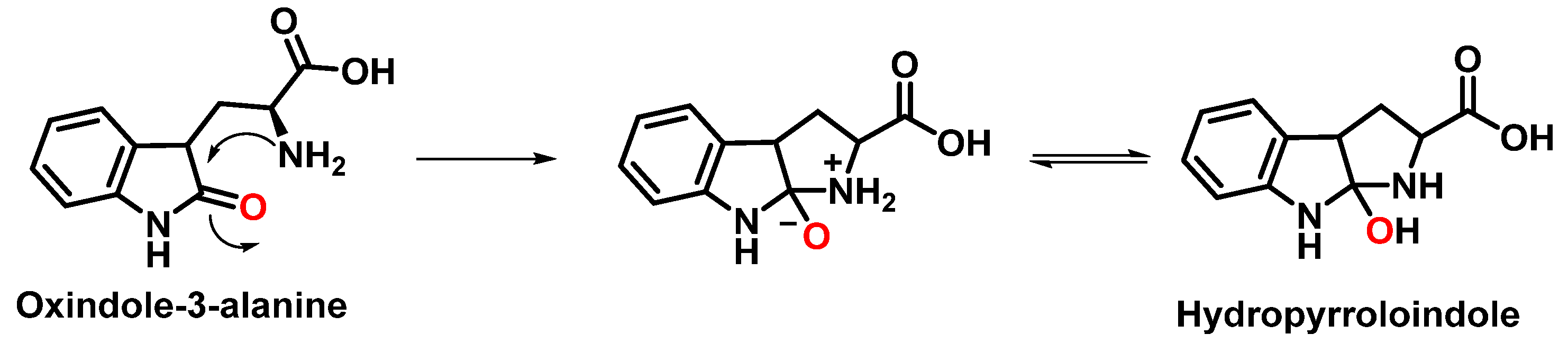
Figure 10. Hydropyrroloindole formation mechanism.
Oxindole-3-alanine, N-formylkynurenine, and four hydroxytryptophans are formed when tryptophan undergoes a Fenton or Udenfriend reaction. According to this observation, the hydroxyl radical targets positions 2 and 3 of the pyrrole ring in addition to the aromatic nucleus. Hydroxylated derivatives are formed when the hydroxyl radical adduct is unevenly distributed with Fe-EDTA, or in a Fenton reaction. The yield of hydroxytryptophane derivatives is proportional to the concentration of Fe-EDTA up to a theoretical yield limit of 54%. The ratio of 4-, 5-, 6-, and 7-hydroxytryptophan derivatives under these circumstances is 4:2:2:3, respectively [21]. Trp subjected to the Udenfriend reaction yields approximately equal amounts of 4-, 5-, 6-, and 7-OHTrp and N-formylkynurenine.
2.3. Cysteine Derivatives from Reaction with Peroxynitrite
Cysteine is the amino acid that reacts most rapidly with peroxynitrite. The products and intermediates detected in the reaction of cysteine with peroxynitrite [22][23] are specified in Figure 11.
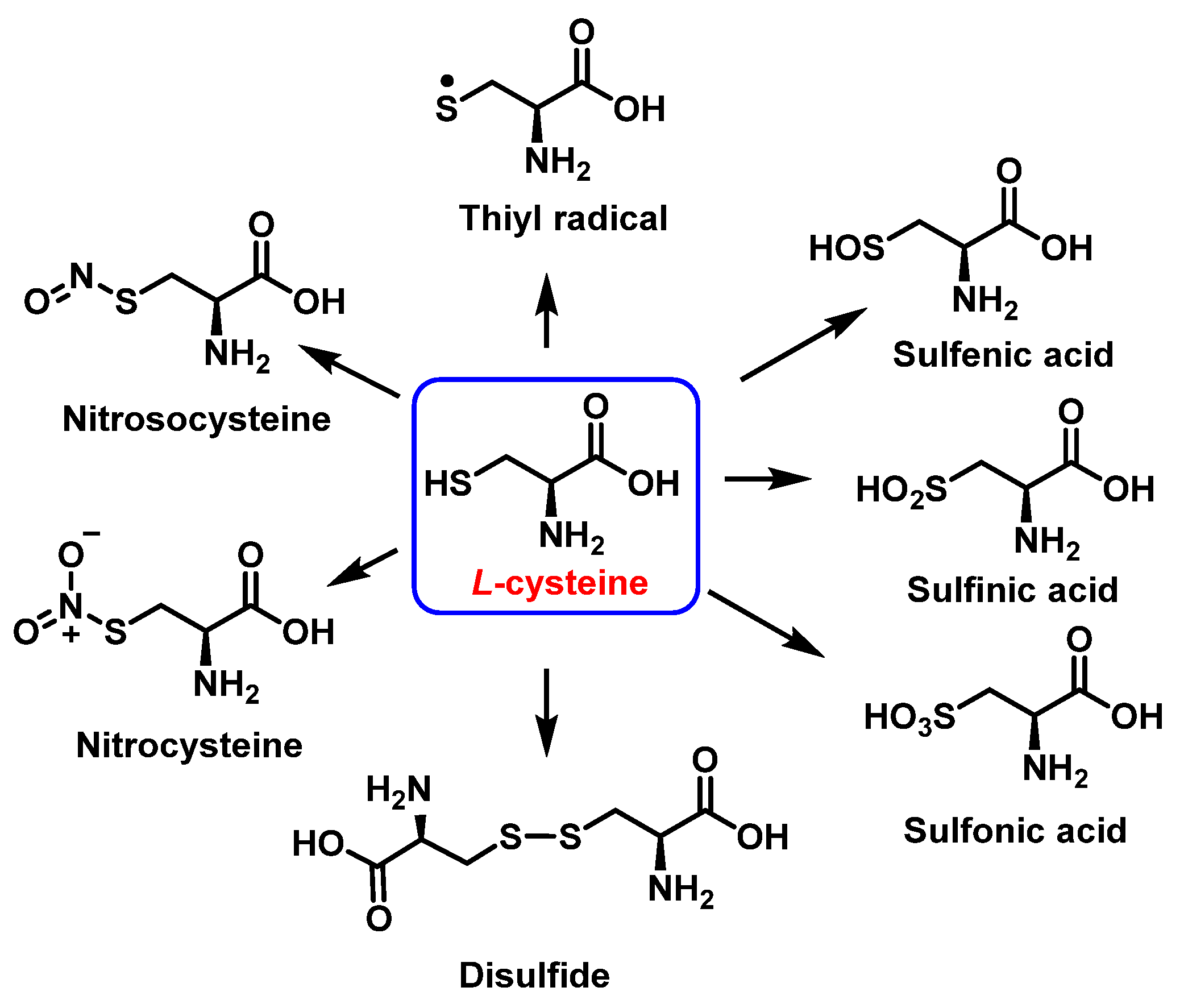
Figure 11. Products and intermediates detected in the reaction of cysteine with peroxynitrite.
In biochemistry, S-Nitrosylation is the covalent attachment of a nitric oxide group −NO to a cysteine thiol within a protein to form an S-nitrosothiol SNO, and has diverse regulatory roles in bacteria, yeast, plants, and in all mammalian cells [24]. It is a fundamental mechanism for cellular signaling across phylogeny, and accounts for the large part of NO bioactivity. S-nitrosylation is a precise, reversible process necessary for a wide range of cell signaling, including, for example, the red blood cell-mediated auto regulation of blood flow, essential for vertebrate life [25]. S-nitrosylation depends on enzyme activity, with three classes of S-nitrosylase enzymes, which operate in concert, analogous to ubiquitinylation [26]. The reverse effect of S-nitrosylation is denitrosylation, also controlled by enzymes. Multiple enzymes have been described and divided into two main classes that mediate denitrosylation of proteins and low molecular weight SNOs, respectively. S-Nitrosoglutathione reductase (GSNOR) is an example of the low molecular weight class [27]. These denitrosylases are involved in the removal of NO from S-nitrosylated cysteine residues, and thus can potentially remodel nitrosative stress under disorder conditions.
2.4. Methionine Derivatives from Reaction with Peroxynitrous Acid and Peroxynitrite
Peroxynitrous acid and peroxynitrite both react with methionine. The corresponding kinetic constants being Kacid = (1.7 ± 0.1) × 103 M−1 s−1 and Kanion = 8.6 ± 0.2 M−1 s−1. Nitrites, nitrates, and methionine sulfoxide were the only byproducts identified, Figure 17.
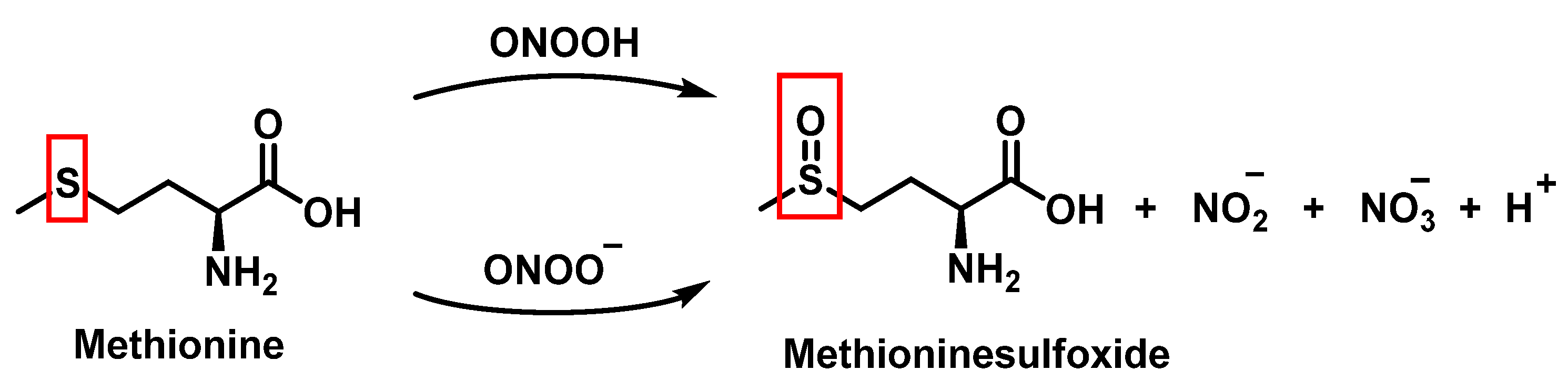
Figure 12. Products and intermediates detected in the reaction of methionine with peroxynitrous acid and peroxynitrite.
For every three peroxynitrites consumed in the reaction, one nitrate and two nitrites are formed. The excess methionine concentration had little effect on the distribution of nitrite, nitrate, and methionine sulfoxide yields. pH had no effect on the production of methionine sulfoxide, nitrite, or nitrate. Methionine sulfoxide with recovered methionine corresponds to 100±4% of the methionine present at the beginning. The only methionine-derived substance identified was methionine sulfoxide [11].
3. Chlorination of Amino Acids
HOCl is formed in biological systems by the enzyme heme myeloperoxidase MPO, which converts H2O2 to HOCl in the presence of chloride ion (Cl−) [28], Figure 13.
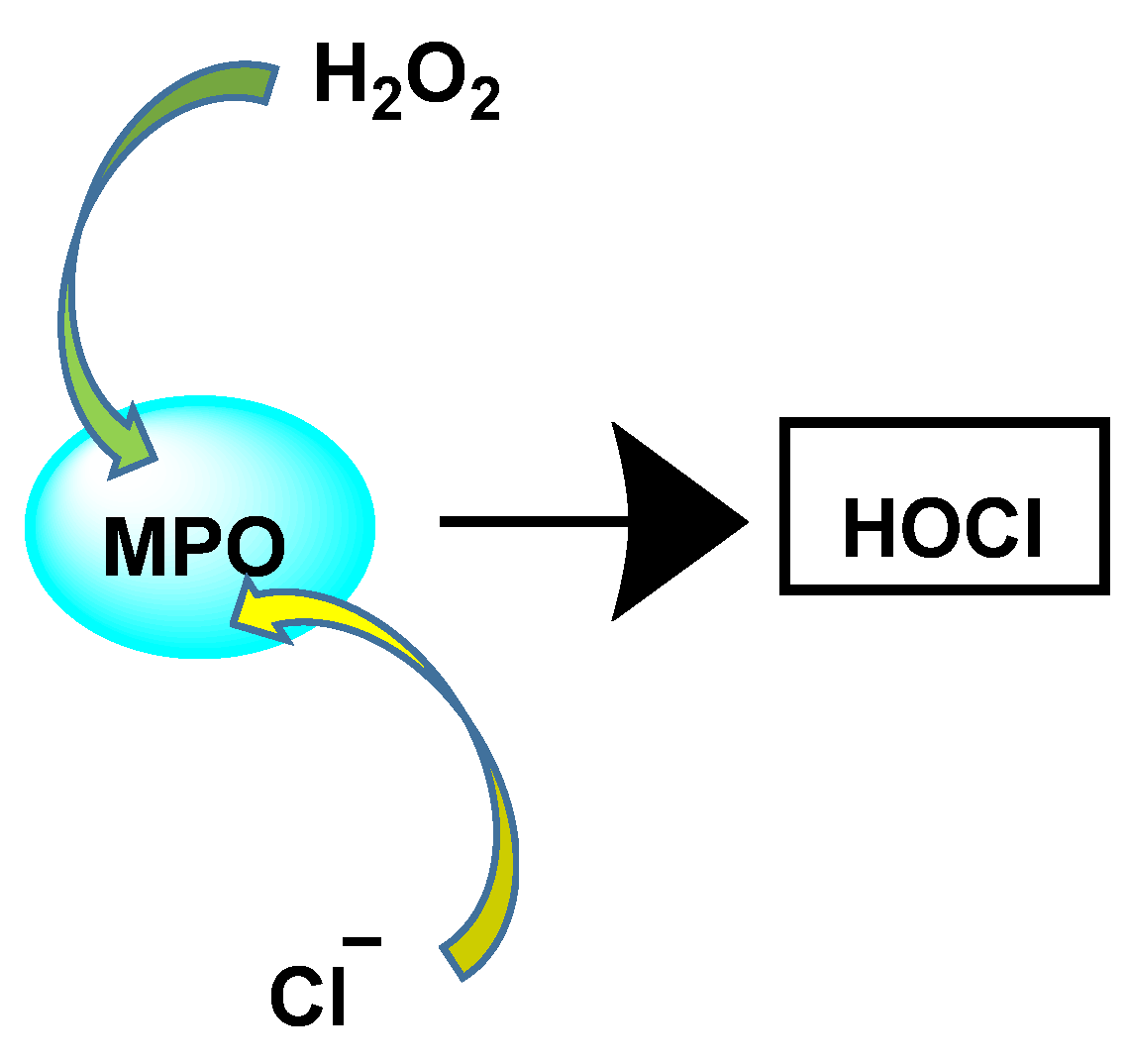
Figure 13. Conversion of H2O2 and chloride to HOCl.
HOCl belongs to a new group of “non-antibiotic antimicrobial molecules”, known as disinfectants, which act by oxidation of organic matter. It was found in very low concentrations in the human body, synthesized by the enzyme myeloperoxidase in the cells of the immune system (neutrophils and macrophages) during an immunological process known as “respiratory burst”, and is used to fight against infections caused by bacteria. It is a chemotactic substance with excellent microbial control. The distance of the H-O bond (97 pm) is almost half that of O-Cl (169.3 pm), so there is a higher electronegativity in the HO part of the molecule, Figure 14. HOCl has a pKa of 7.5, so it coexists with ionized hypochlorite (-OCl) in solution at physiological pH. The HOCl produced has been shown to be a potent oxidant capable of chlorinating electron-rich substrates.
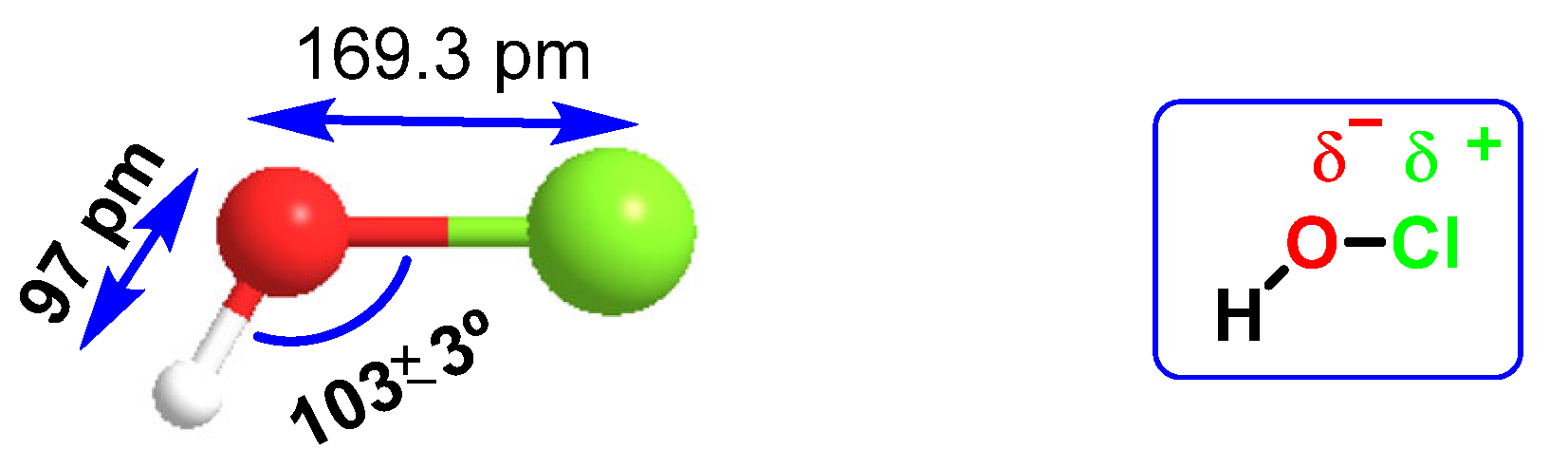
Figure 14. Interatomic distances and electronegativity of the HOCl molecule.
Amino acids can react with HOCl to monochloramine formation, Figure 15. Aromatic amino acids react with HOCl to form a short-lived chloro-amine, which rapidly transfers its chlorine group to another amine.
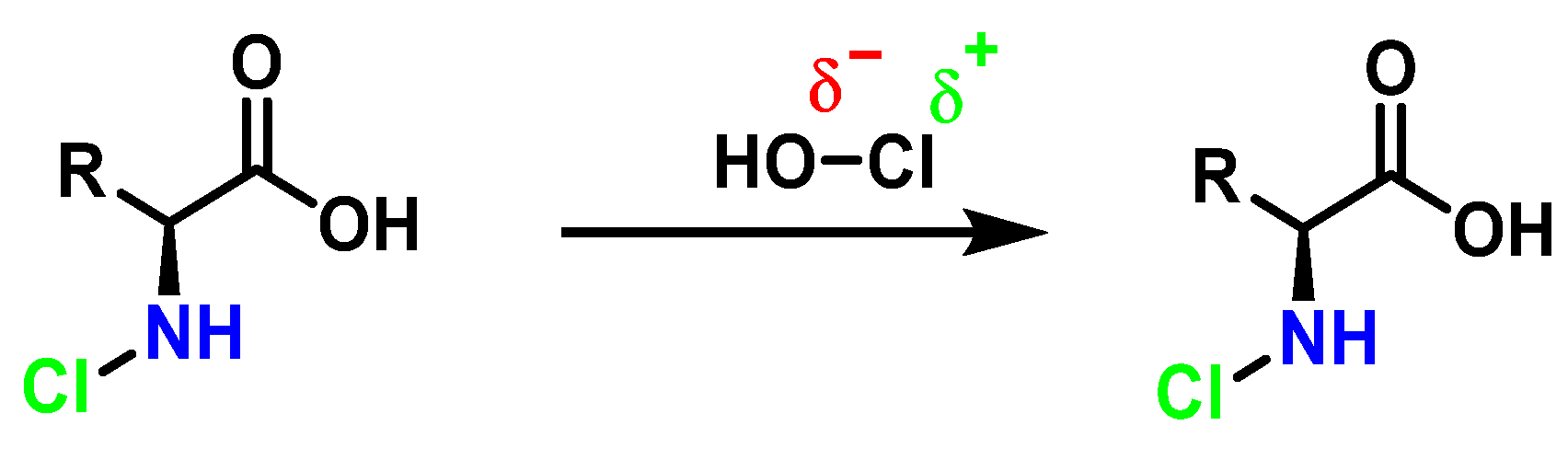
Figure 15. General method of chlorination of the amino group of the amino acid.
Competitive kinetic studies and stopped-flow approaches have been used to evaluate the rates of various HOCl-amino acid reactions [29]. Winterbourn used a competitive reaction with monochlorodimetone in 1985 to determine the relative reaction rates of various amino acids with HOCl. The reaction sites (amino versus side chain) were not reported, although the order of reactivity was Cys > Met > Cystine > His > Ser > Leu.
Second-order rate constants for HOCl reactions within a protein were calculated (at physiological pH, 7.4, in aqueous solution). Met > Cys > Cystine > His > Trp > Lys > Tyr > Arg > Gln > Asn [30] was the order of reactivity of the different side chains. The constants for the reactions of HOCl with the side-chain groups of amino acids, α--amino groups, and backbone amides are shown.
Depending on the environment, the second-order rate constant for backbone amides varies by several orders of magnitude. This is a maximum estimate based on research with cyclic dipeptides.
In view of the energy content of the chlorinated species in the amino acids studied, a little higher than the free amino acids, it can be proposed that chlorination takes place, but that the new molecule evolves by losing chlorine as a radical, or through decarboxylation of the chloramines, to form unstable imines that undergo hydrolysis and change to aldehydes, with loss of ammonia.
It was now considered that the products and energy content of the side-chain reaction on a series of amino acids with HOCl, Figure 16.
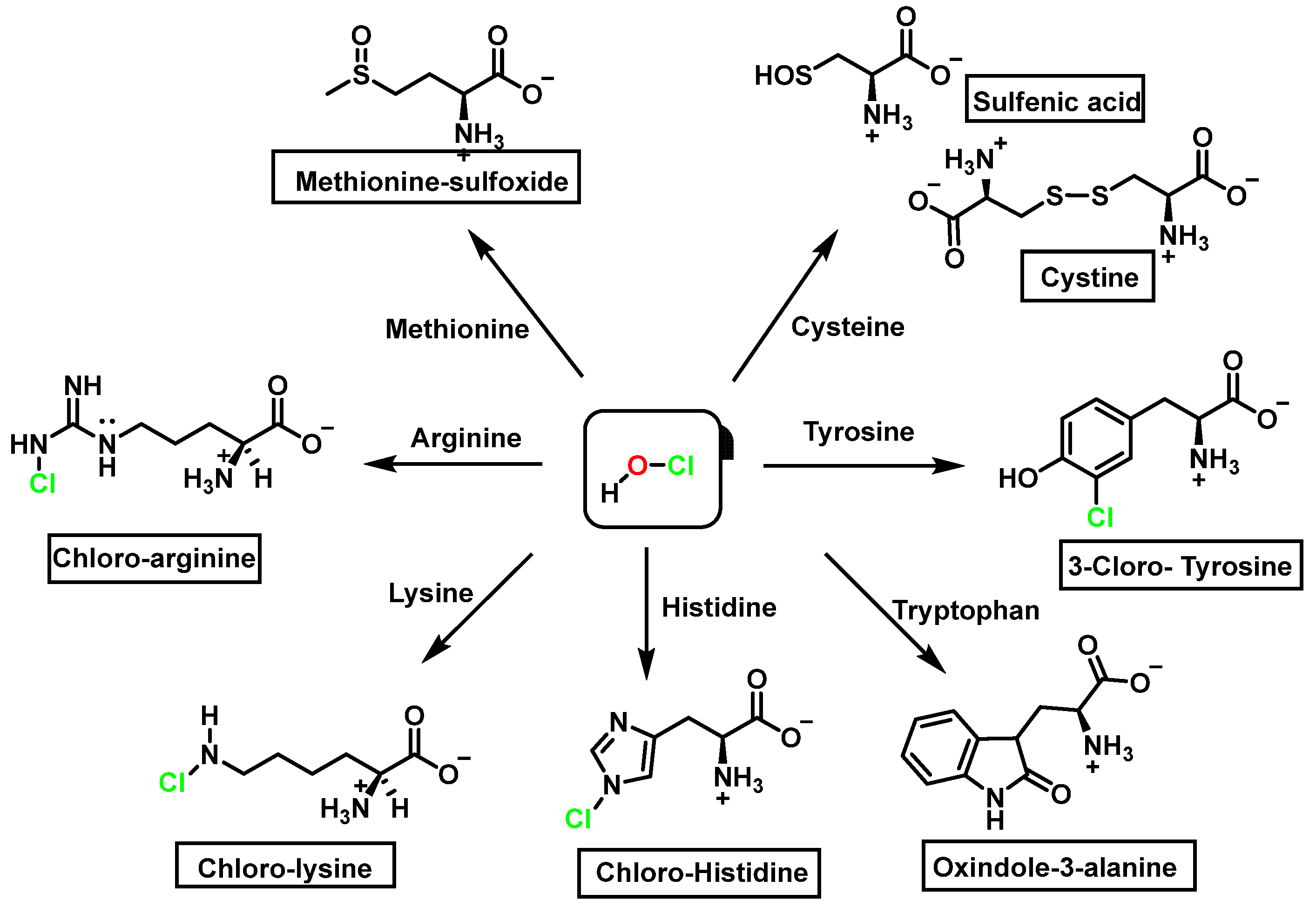
Figure 16. Chlorination of amino acids to a nitrogen other than the amino group adjacent to the carboxyl group.
Reaction with HOCl alters the side chains of tyrosine, tryptophan, histidine, arginine, cysteine, and methionine derivatives. The main products of HOCl-mediated Trp oxidation are 2-hydroxyindole and its tautomeric 2-oxindole derivative Trp [31]. It has also been proposed that HOCl oxidation of Trp can lead to the formation of kynurenine and N-formylkynurenine. The proposed mechanism involves HOCl reacting with the amino group to form monochloramine, which is then thermally decomposed (or catalyzed by metal ions) to form radicals. These radicals can react with additional Trp side chains to form kynurenine.
In the presence of excess HOCl, the reactions of the amine of the Lys side chain with HOCl lead to the formation of unstable mono- and dichloramines. These compounds are moderate oxidants that can transfer chlorine to other substrates while the original amine is renewed [32].
Although the His side chain reacts rapidly with HOCl to form a short-lived chloramine [33], the free amino group can also be chlorinated. At a pH of 7.4, kinetic measurements indicate that the reaction proceeds approximately the same at both sites [30]. However, because the reactivity of the imidazole ring is particularly sensitive to pH, changes in pH, an environment in which the pKa of the side chain changes, limit the rate of reaction at the side chain. This leads to a preferential reaction at the -amino group. Ring-derived chloramines have been shown to readily transfer chlorine to other amine groups, resulting in more stable chloramines. The interaction of histamine with HOCl at pH 8.0 has already been shown to occur preferentially at the (non-imidazole) amino main group [34].
The aromatic ring reaction of tyrosine with HOCl leads to 3-chlorotyrosine. There is no information in the literature about the by-products of HOCl-mediated oxidation of Gln and Asn side chains in aqueous solution. This is most likely due to their slow reaction rate with HOCl, which makes them very insignificant targets in proteins [30].
References
- Kurutas, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr. J. 2015, 15, 71.
- Zarkovic, N. Roles and Functions of ROS and RNS in Cellular Physiology and Pathology. Cells 2020, 9, 767.
- Shields, H.J.; Traa, A.; Van Raamsdonk, J.M. Beneficial and detrimental effects of reactive oxygen species on lifespan: A comprehensive review of comparative and experimental studies. Front. Cell Dev. Biol. 2021, 9, 628157.
- Khairutdinov, R.F.; Coddington, J.W.; Hurst, J.K. Permeation of phospholipid membranes by peroxynitrite. Biochemistry 2000, 39, 14238–14249.
- Bandookwala, M.; Sengupta, P. 3-Nitrotyrosine: A versatile oxidative stress biomarker for major neurodegenerative diseases. Int. J. Neurosci. 2020, 130, 1047–1062.
- Yamakura, F.; Matsumoto, T.; Ikeda, K.; Taka, H.; Fujimura, T.; Murayama, K.; Watanabe, E.; Tamaki, M.; Imai, T.; Takamori, K. Nitrated and oxidized products of a single tryptophan residue in human Cu, Zn-superoxide dismutase treated with either peroxynitrite-carbon dioxide or myeloperoxidase-hydrogen peroxide-nitrite. J. Biochem. 2005, 138, 57–69.
- Ishii, Y.; Ogara, A.; Katsumata, T.; Umemura, T.; Nishikawa, A.; Iwasaki, Y.; Ito, R.; Saito, K.; Hirose, M.; Nakazawa, H. Quantification of nitrated tryptophan in proteins and tissues by high-performance liquid chromatography with electrospray ionization tandem mass spectrometry. J. Pharm. Biomed. Anal. 2007, 44, 150–159.
- Hernansanz-Agustín, P.; Izquierdo-Alvarez, A.; García-Ortiz, A.; Ibiza, S.; Serrador, J.M.; Martínez-Ruiz, A. Nitrosothiols in the immune system: Signaling and protection. Antioxid. Redox Signal. 2013, 18, 288–308.
- Wijasa, T.S.; Sylvester, M.; Brocke-Ahmadinejad, N.; Schwartz, S.; Santarelli, F.; Gieselmann, V.; Klockgether, T.; Brosseron, F.; Heneka, M.T. Quantitative proteomics of synaptosome S-nitrosylation in Alzheimer’s disease. J. Neurochem. 2020, 152, 710–726.
- Mishra, D.; Patel, V.; Banerjee, D. Nitric oxide and S-nitrosylation in cancers: Emphasis on breast cancer. Breast Cancer Basic Clin. Res. 2020, 14, 1178223419882688.
- Perrin, D.; Koppenol, W.H. The quantitative oxidation of methionine to methionine sulfoxide by peroxynitrite. Arch. Biochem. Biophys. 2000, 377, 266–272.
- Nakao, L.S.; Iwai, L.K.; Kalil, J.; Augusto, O. Radical production from free and peptide-bound methionine sulfoxide oxidation by peroxynitrite and hydrogen peroxide/iron (II). FEBS Lett. 2003, 547, 87–91.
- Chapman, A.L.; Winterbourn, C.C.; Brennan, S.O.; Jordan, T.W.; Kettle, A.J. Characterization of non-covalent oligomers of proteins treated with hypochlorous acid. Biochem. J. 2003, 375, 33–40.
- Radi, R. Protein tyrosine nitration: Biochemical mechanisms and structural basis of functional effects. Acc. Chem. Res. 2013, 46, 550–559.
- Boron, W.F. Regulation of intracellular pH. Adv. Physiol. Educ. 2004, 28, 160–179.
- Nuriel, T.; Whitehouse, J.; Ma, Y.; Mercer, E.J.; Brown, N.; Gross, S.S. ANSID: A solid-phase proteomic approach for identification and relative quantification of aromatic nitration sites. Front. Chem. 2016, 3, 70.
- Koppenol, W.; Moreno, J.; Pryor, W.A.; Ischiropoulos, H.; Beckman, J. Peroxynitrite, a cloaked oxidant formed by nitric oxide and superoxide. Chem. Res. Toxicol. 1992, 5, 834–842.
- Butler, J.; Land, E.J.; Prütz, W.A.; Swallow, A.J. Charge transfer between tryptophan and tyrosine in proteins. Biochim. Biophys. Acta (BBA) Protein Struct. Mol. Enzymol. 1982, 705, 150–162.
- Alvarez, B.; Radi, R. Peroxynitrite reactivity with amino acids and proteins. Amino Acids 2003, 25, 295–311.
- Kato, Y.; Kawakishi, S.; Aoki, T.; Itakura, K.; Osawa, T. Oxidative modification of tryptophan residues exposed to peroxynitrite. Biochem. Biophys. Res. Commun. 1997, 234, 82–84.
- Maskos, Z.; Rush, J.D.; Koppenol, W.H. The hydroxylation of tryptophan. Arch. Biochem. Biophys. 1992, 296, 514–520.
- Van der Vliet, A.; Hoen, P.A.; Wong, P.S.; Bast, A.; Cross, C.E. Formation of S-nitrosothiols via direct nucleophilic nitrosation of thiols by peroxynitrite with elimination of hydrogen peroxide. J. Biol. Chem. 1998, 273, 30255–30262.
- Balazy, M.; Kaminski, P.M.; Mao, K.; Tan, J.; Wolin, M.S. S-Nitroglutathione, a product of the reaction between peroxynitrite and glutathione that generates nitric oxide. J. Biol. Chem. 1998, 273, 32009–32015.
- Stomberski, C.T.; Hess, D.T.; Stamler, J.S. Protein S-nitrosylation: Determinants of specificity and enzymatic regulation of S-nitrosothiol-based signaling. Antioxid. Redox Signal. 2019, 30, 1331–1351.
- Marozkina, N.V.; Gaston, B. S-Nitrosylation signaling regulates cellular protein interactions. Biochim. Biophys. Acta (BBA) Gen. Subj. 2012, 1820, 722–729.
- Anand, P.; Stamler, J.S. Enzymatic mechanisms regulating protein S-nitrosylation: Implications in health and disease. J. Mol. Med. 2012, 90, 233–244.
- Stomberski, C.T.; Zhou, H.-L.; Wang, L.; van den Akker, F.; Stamler, J.S. Molecular recognition of S-nitrosothiol substrate by its cognate protein denitrosylase. J. Biol. Chem. 2019, 294, 1568–1578.
- Pattison, D.I.; Davies, M.J. Reactions of myeloperoxidase-derived oxidants with biological substrates: Gaining chemical insight into human inflammatory diseases. Curr. Med. Chem. 2006, 13, 3271–3290.
- Winterbourn, C.C. Comparative reactivities of various biological compounds with myeloperoxidase-hydrogen peroxide-chloride, and similarity of the oxidant to hypochlorite. Biochim. Biophys. Acta 1985, 840, 204–210.
- Pattison, D.I.; Davies, M.J. Absolute rate constants for the reaction of hypochlorous acid with protein side chains and peptide bonds. Chem. Res. Toxicol. 2001, 14, 1453–1464.
- Joule, J.A.; Mills, K.; Smith, G.F. Heterocyclic Chemistry; CRC Press: Boca Raton, FL, USA, 2020.
- Thomas, E.L.; Grisham, M.B.; Jefferson, M.M. Preparation and characterization of chloramines. Methods Enzym. 1986, 132, 569–585.
- Thomas, E.L.; Jefferson, M.M.; Bennett, J.J.; Learn, D.B. Mutagenic activity of chloramines. Mutat. Res. 1987, 188, 35–43.
- Thomas, E.L.; Jefferson, M.M.; Learn, D.B.; King, C.C.; Dabbous, M.K. Myeloperoxidase-catalyzed chlorination of histamine by stimulated neutrophils. Redox Rep. 2000, 5, 191–196.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.1K
Entry Collection:
Chemical Bond
Revisions:
2 times
(View History)
Update Date:
23 Nov 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No