 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ágnes Ódri | + 2033 word(s) | 2033 | 2020-11-16 10:56:00 | | | |
| 2 | Ágnes Ódri | Meta information modification | 2033 | 2020-12-15 12:26:29 | | | | |
| 3 | Ágnes Ódri | Meta information modification | 2033 | 2020-12-15 12:30:25 | | |
Video Upload Options
Definition
Acid rock drainage (ARD) is one of the major environmental problems at active and closed coal and metalliferous mining sites. ARD forms naturally when sulfidic material comes in contact with atmospheric oxygen and the oxidation of sulfide minerals releases acid, sulfate and metal contaminants. The large-scale ARD that is generated within the mining area is called acid mine drainage (AMD).
Introduction
The characterization of acid rock drainage (ARD) is traditionally based on mineralogical and geochemical techniques (e.g., Acid Base Accounting tests). The complexity of ARD processes warrants contribution of methods from various disciplines. In the past decade, the increasing role of environmental isotopes in pollution monitoring has enabled the successful application of isotope methods in ARD investigations. While isotopic compositions of different pollutants can refer to their parent mineral, the degree of isotope fractionations are indicative of the mechanisms taking place during the release and transportation of ARD-related contaminants. In natural environments, however, the measured isotope fractionations are predominantly the result of several coexisting or sequential processes. Therefore, the identification and quantification of the distinct contributions of these processes to isotope variations is difficult and requires well-defined laboratory conditions, where the influence of ARD generation on different isotope systems can be assessed with greater certainty.
Sulfur Isotope Signatures of Pyrite Leaching
Sulfur isotope composition of dissolved sulfates in ARD—generated via Reactions (1) and (3)—relates to the source of sulfur, therefore it has the potential to be successfully used in mass balance calculations as part of the pollution monitoring: [1]
FeS2 + 3.5O2 + H2O → Fe2+ + 2SO42- + 2H+ (1)
Fe2+ + H+ + 0.25O2 → Fe3+ + 0.5H2O (2)
FeS2 + 14Fe3+ + 8H2O → 15Fe2+ + 2SO42- + 16H+ (3)
Pyrite leaching experiments demonstrate that the quantitative conversion of pyrite to sulfate results in only minor sulfur isotope fractionation (Δ34SSO4–FeS2) under acidic, aerobic-biotic, or abiotic conditions ranging between −1.3‰ and +0.4‰.[2][3][4][5][6][7][8] In addition, processes like precipitation and dissolution of secondary sulfate phases were also demonstrated to produce negligible S isotope fractionation (e.g., Δ34Sschwertmannite–sulfate −0.2‰ – +0.3‰ and Δ34SH-jarosite–sulfate +0.3‰ – +1.1‰).[9][10][11] Although these relatively small S isotope fractionation factors allow the identification of the parent sulfides in ARD-impacted water, the seasonal precipitation and dissolution cycles of sulfate minerals might cause small variations in the isotopic composition of sulfate. According to the overall stoichiometry of pyrite oxidation mechanisms, abiotic oxidation (Reaction (1)) can take place only when there is enough dissolved oxygen to complete the reaction. Even though Reactions (1) and (3) are the most frequently used pathways to demonstrate pyrite oxidation, the actual mechanisms are much more complex and involve the occurrence of intermediate sulfur species like thiosulfate (S2O32−), elemental sulfur (So), and sulfite (SO32−), depending on the availability of oxygen. Pyrite is mostly oxidized by bacterially generated Fe3+ (Reaction (3)) via thiosulfate oxidation pathway.[12] The formation of sulfur intermediates during abiotic and biotic pyrite oxidation was confirmed by several authors both under acidic and alkaline conditions, reporting systematically smaller amounts of intermediates at lower pH due to their rapid oxidation to sulfate.[13][14][15][16][17][18][19][20][21][22][23] The incomplete oxidation of pyrite or its stepwise oxidation to sulfate is frequently accompanied by sulfur isotope variation in agreement with the general concepts of isotope fractionation, and the more oxidized form of sulfur is enriched in the heavier isotope compared to more reduced forms.[24] As S and O isotopes of sulfate are intensively involved in redox reactions that control the sulfur oxyanion formations, the isotope fractionation has the potential to provide insight on the governing pathways of sulfur oxidation or reduction. Due to the complexity of these redox processes along with the large number of variables that control the sulfur conversion mechanisms (e.g., residence time of sulfur intermediates, pH, crystal structure of the leached mineral, bacterial occurrence, oxygen availability, dominant oxidant), current understanding of the influence of sulfur intermediates on both the ARD sulfur cycle dynamics and its isotope geochemical characteristics is relatively limited.
Oxygen Isotope Signatures of Pyrite Leaching
Sulfate preserves its oxygen isotope composition after formation. This allows for the identification of the reaction mechanisms that are responsible for sulfide oxidation; if most of the sulfate oxygen is derived from the dissolved molecular oxygen via Reaction (1), the emerging SO4 should be enriched in 18O relative to SO4 that incorporates dominantly water-derived oxygen (WDO) following Reaction (3). The reason for this is that air is characterized by δ18O = +23.8 ‰,[25] whereas meteoric water has variable δ18O values mainly δ18O ≤ 0 ‰.[26] Using Equation (4), the relative contributions of WDO and molecular oxygen to dissolved sulfate can be calculated.[27]
δ18OSO4 = X(δ18OH2O + εH2O) + (1 – X) (δ18OO2 + εO2) (4)
where X is the fraction of sulfate produced by Reaction (3) and (1 – X) is the fraction of sulfate from Reaction (1). ε is the enrichment factor that describes the partition of oxygen between the sulfate and air or water. ε18OSO4-H2O = 0.0 ‰ to +4.1 ‰ for both anaerobic biotic and abiotic experiments and ε18OSO4-O2 = -10.0 ‰ to -11.4 ‰ for biotic and -4.3 ‰ to -9.8 ‰ for abiotic reactions.[2][5][27]
The general concept regarding the relative percent contribution of various oxygen sources to sulfate is that: (i) if pyrite oxidation is anaerobic and Reaction (3) is the dominant, then 100% of the sulfate oxygen is derived from water and (ii) if dissolved molecular oxygen is present, sulfate oxygen shows a combination of air and water oxygen. Pyrite oxidation Reaction (1) suggests that 87.5% of sulfate oxygen is dissolved molecular oxygen and the remaining 12.5% is water derived. The WDO values, however, do not necessarily reflect the stoichiometry of Reactions (1).[2][5] To understand the reason for significant water oxygen incorporation into sulfate at low pH even under oxygen-saturated conditions, it is necessary to review the mechanism of pyrite oxidation. The initial step is the oxidation of the adsorbed Fe2+(H2O)6 complexes on the pyrite surface by molecular oxygen to form Fe3+(H2O)6 complexes that rapidly oxidize the pyrite (Figure 1).[14][21] During the initial release of Fe2+ to solution, the pyrite surface is dominated by sulfur atoms. The adsorbed iron continuously experiences oxidation and reduction during the interaction with molecular oxygen and the acceptance of electrons from the pyrite. The latter process is governed by the electron loss of iron on the pyrite surface during the interaction via molecular oxygen.[13] The electron originates from the sulfur site and its transmission to Fe3+ takes place via the iron site of the mineral (Figure 1b).
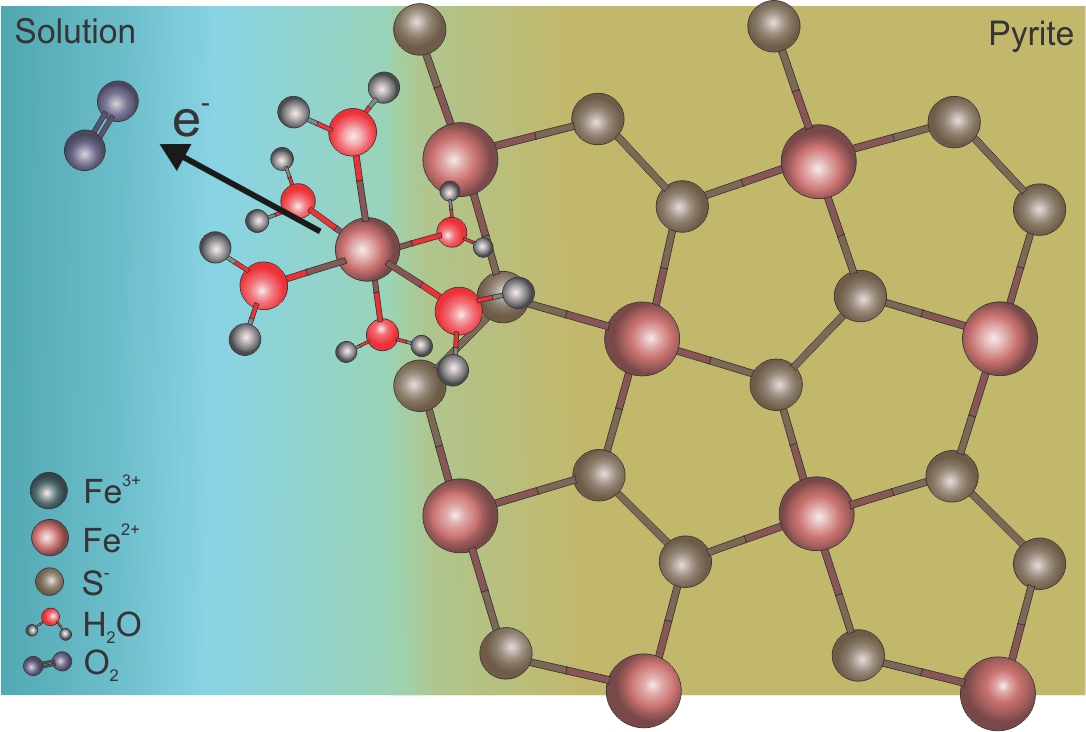
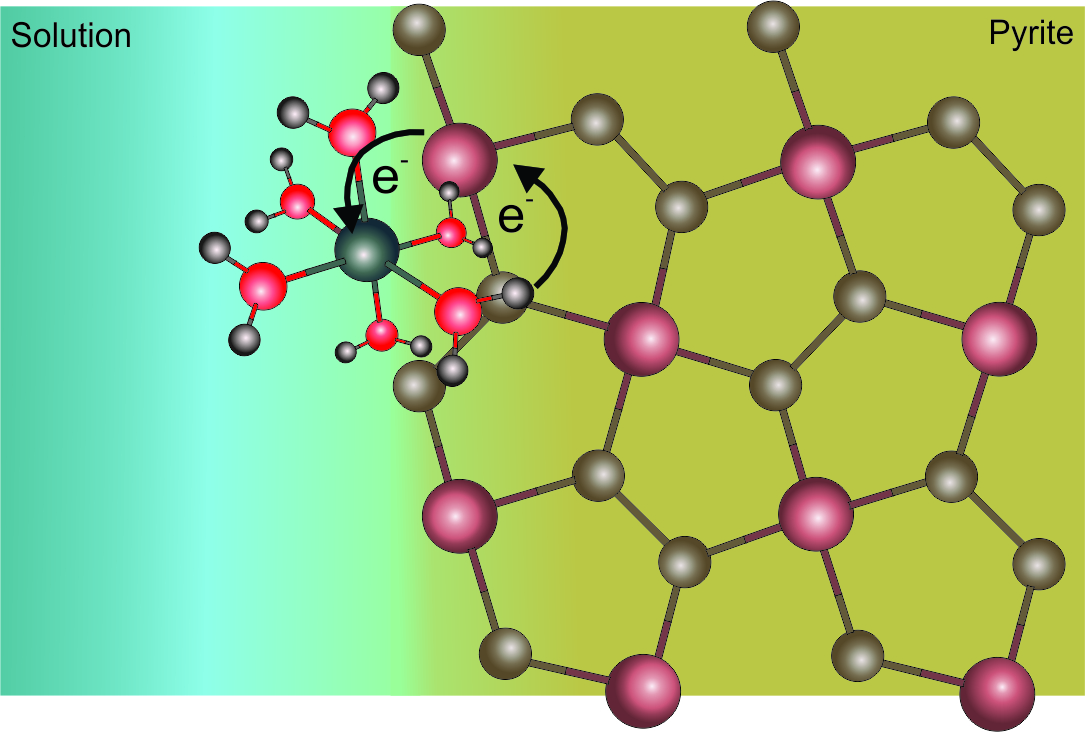
(a) (b)
Figure 1. Illustration of the abiotic oxidation mechanism of pyrite. The adsorption of released Fe2+ from pyrite as aqueous Fe2+(H2O)6 is the first step of pyrite oxidation. (a) The oxidation of Fe2+(H2O)6 complex by the dissolved molecular oxygen forms (b) Fe3+ that accepts electron from the pyrite during its oxidation.
An oxygen atom from the adsorbed Fe3+(H2O)6 complex is added to the sulfur site of pyrite as the closing step of pyrite oxidation that produces different sulfur intermediates (Figure 2a).[19][22] The reaction sequence of the oxidation-reduction of the Fe3+ in the adsorbed aqueous complex, and the transfer of oxygen from water molecules to the sulfur site of pyrite, continues until a sulfur oxyanion is more stable in solution, and disassociates from the pyrite surface (Figure 2b).[14]
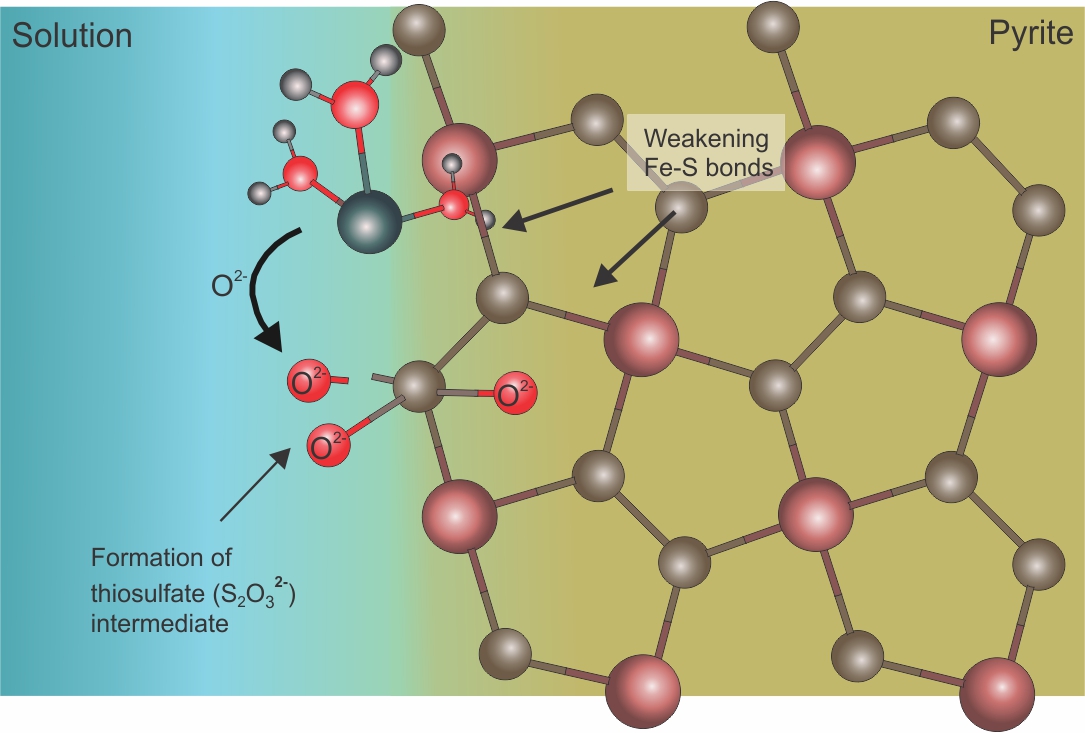
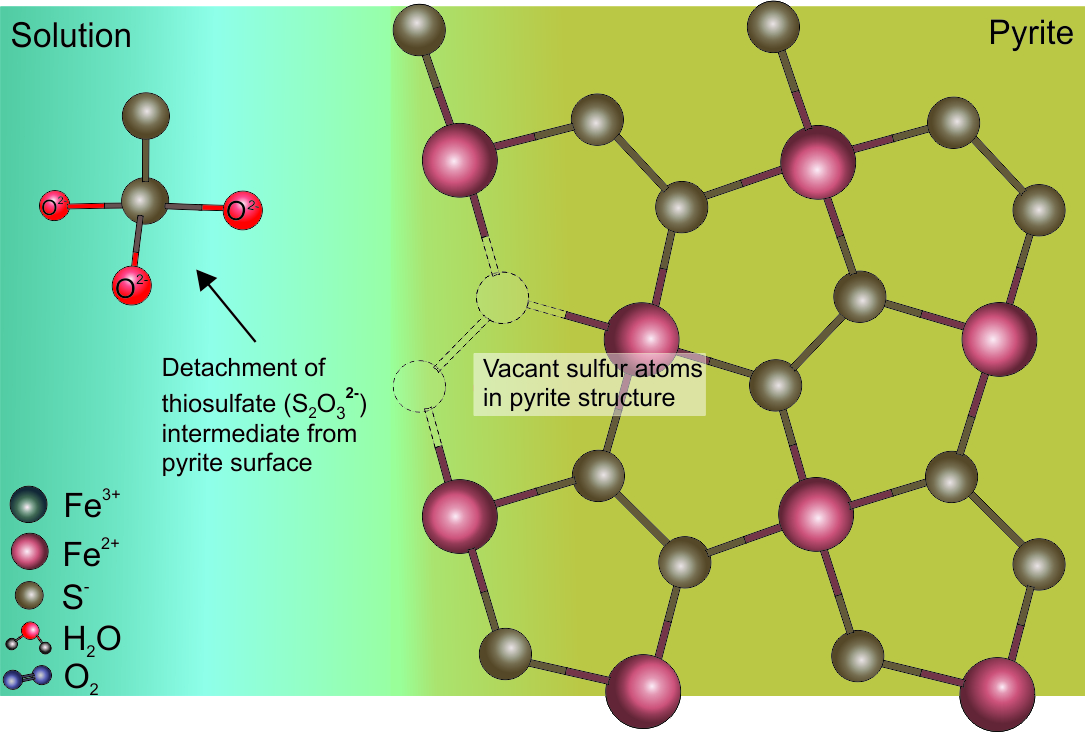
(a) (b)
Figure 2. Illustration of the formation of sulfur oxyanion during the abiotic oxidation of pyrite using the example of thiosulfate. (a) Addition of oxygen molecules from the aqueous Fe3+(H2O)6 complex to pyrite sulfur to form thiosulfate. (b) The oxygen transfer continues until the detachment of the stable thiosulfate.
In accordance with the mechanism described above, oxygen plays a role in the initiation of the pyrite oxidation reaction, but the reaction pathway is mainly controlled by Fe3+ as the oxidant. As molecular oxygen is not directly involved in the formation of sulfur intermediates, the incorporation of water oxygen into thiosulfate from Fe3+(H2O)6 complexes is the major process that affects the oxygen isotope composition of this sulfur compound. The participation of molecular oxygen in sulfate takes place at the more advanced stages of oxidation. The major potential factors that are considered to cause variations in sulfate oxygen isotope values and the ratio of WDO and O2 are: (i) the contribution of different oxygen sources, (ii) degree of the oxygen isotope exchange between water and various sulfur intermediates, (iii) environmental conditions such as pH or availability of oxygen and (iv) the reaction mechanisms of pyrite oxidation through the related sulfur intermediates.[20][28]
Iron Isotope Signatures of Pyrite Leaching
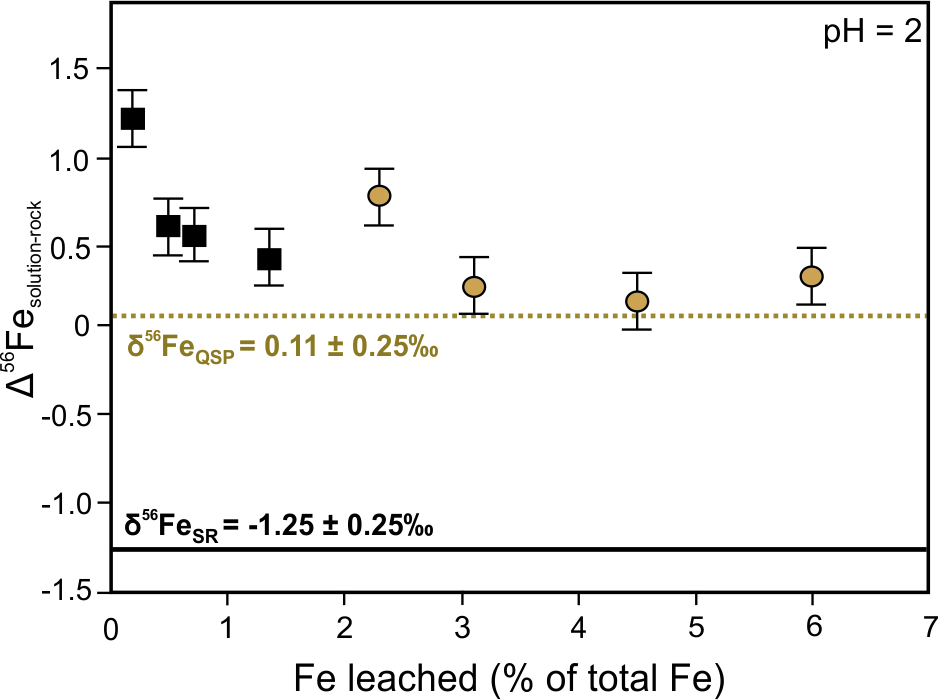
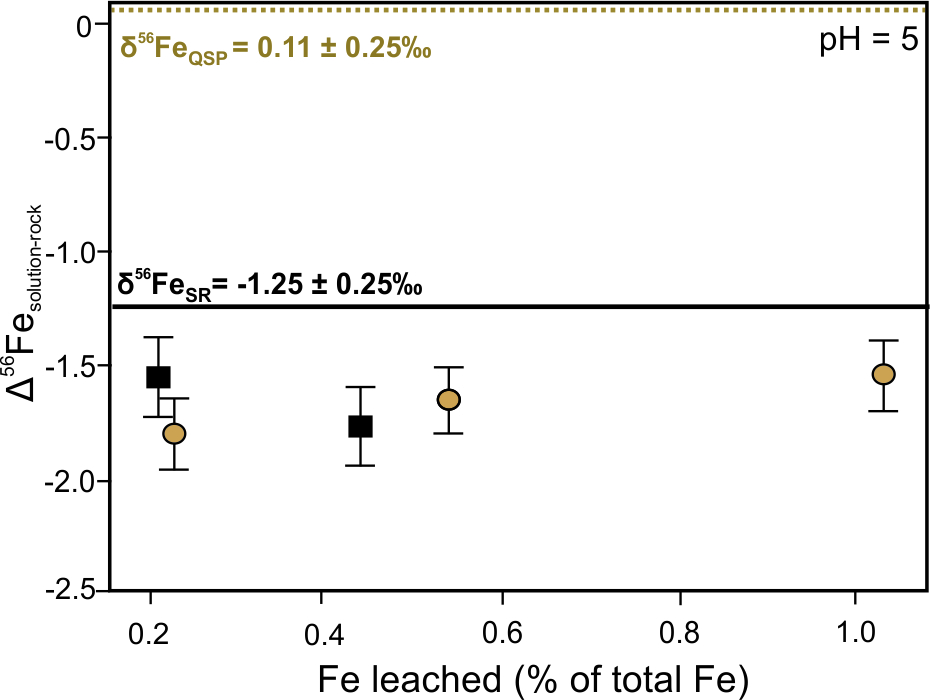
Laboratory experiments have demonstrated isotope fractionation from 2.63 ‰ to 3.00 ‰ between coexisting aqueous Fe(II) and Fe(III) complexes and precipitated Fe(III) phases.[29][30] The degree of Fe isotope fractionation during Fe(III) oxides and (oxy)hydroxides precipitation is significantly affected by factors such as grain size and the rate of precipitation in addition to pH conditions.[31] The enrichment of 56Fe in the fluid phase at the start of oxidative dissolution of sulfide-rich rocks at pH 2 has been demonstrated (Figure 3a).[32] The initial enrichment was caused by the dissolution of a 56Fe-rich, thin layer of ferric-oxide on the sulfide surface that was the result of air oxidation prior to leaching This transient redox isotope effect was followed by the dominance of kinetic fractionation as more iron was released with time, which decreased the δ56Fe value of the solution. Figure 3b shows that during the dissolution of sulfide-rich rocks under circumneutral conditions, the leachate is consistently characterized by lower δ56Fe values compared to bulk rock due to the immediate formation of secondary precipitates.[32] Whereas the aqueous oxidation of Fe2+ under acidic abiotic conditions (pH 2)—described by Reaction (2)—is limited due to its sluggish kinetics,[33] the rapid oxidation of aqueous Fe(II) and the subsequent precipitation of Fe(III) (oxy)hydroxides is enabled at circumneutral pH values (pH 5). The precipitated Fe(III) prefers the incorporation of heavy Fe isotopes, that results in a decrease of the Fe isotope ratios in the fluid phase (Figure 3b).
(a) (b)
Figure 3. Fractionation of iron isotopes between solution and sulfide-rich rocks (Δsolution-rock = δ56Fesolution – δ56Ferock) as a function of Fe recovery (a) under pH = 2 and (b) pH = 5 conditions during continuous batch leaching. Square symbols—leachate of sulfide rock (SR) group: pyrite, chalcopyrite, galena, sericite, pyrophyllite, anglesite, sphalerite. δ56FeSR = –1.25 ± 0.25‰. Circle symbols—leachate of quartz-sericite-pyrite (QSP) group: pyrite, sericite, pyrophyllite, quartz. δ56FeQSP = 0.11 ± 0.25‰. Redrawn from Fernandez and Borrok.[32]
Influence of Microorganisms on Iron Isotope Fractionation
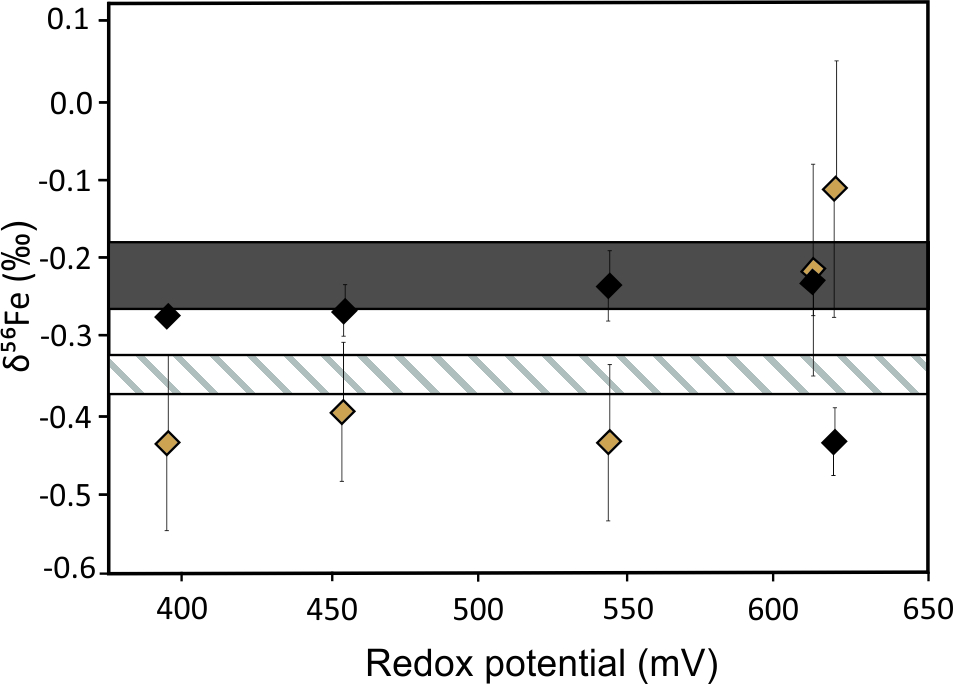
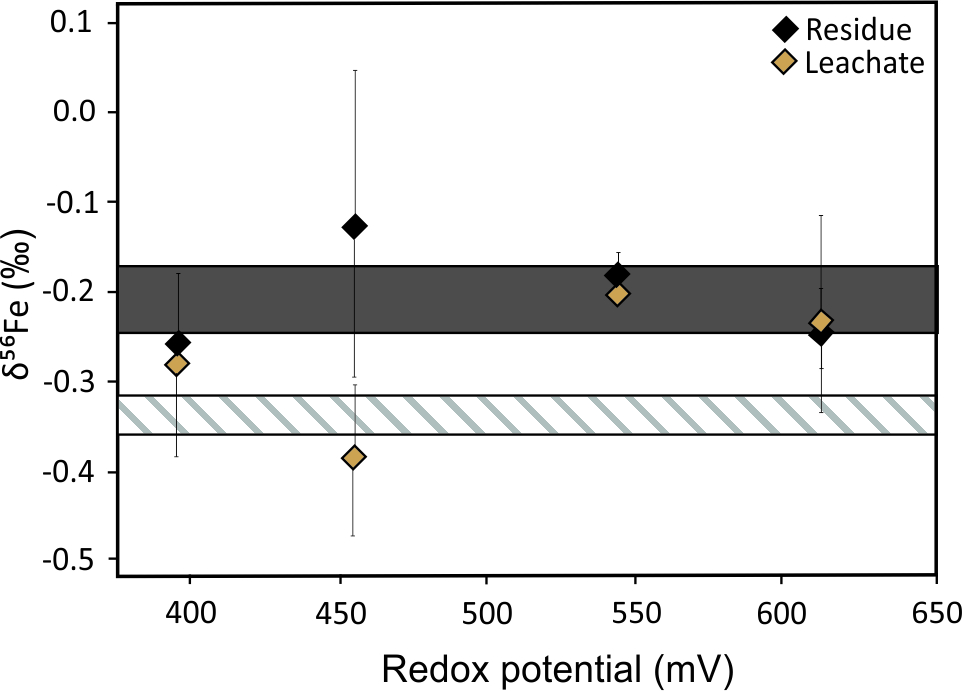
Leaching experiments of different iron minerals demonstrate both the presence and absence of bacterial control on Fe isotopes during the dissolution. The authors of Croal et al.[34] demonstrated 1.5‰ microbial fractionation between aqueous Fe(II) and Fe(III). This suggests the biological control on redox processes and subsequent isotope fractionations. In contrast, the authors of Balci et al.[35] identified abiotically controlled Fe isotope fractionation between Fe(II) and Fe(III) under acidic conditions (pH < 3). Bio- and electrochemical leaching experiments of metal sulfides under consistent redox conditions found no significant difference in Fe isotope fractionation (Figure 4).[36] This emphasizes the importance of redox potential conditions as the main factor governing the redistribution of Fe isotopes during sulfide dissolution, which seems to be a unique signature of the sulfide system.
(a) (b)
Figure 4. Plot of δ56Fe values in residues and leachates as a function of redox potential for (a) bioleaching and (b) electrochemical leaching of pyritic chalcopyrite concentrate. Grey areas represent the initial isotopic composition of the mineral concentrate with 2σ errors. Dashed areas represent the initial isotopic composition of the solution with 2σ errors. Error bars of samples are 2σ. Redrawn from Pérez Rodríguez et al.[36]
Concordant Fe isotope variation mechanisms in biotic and abiotic experiments do not allow for the distinction between biological Fe fractionation and that of abiotic chemical reactions, and points to the complexity of controlling mechanisms of various isotope systems even under laboratory conditions. Further investigations that allow for the accurate characterization of abiotic and biotic processes are warranted.
Summary
The importance of the oxidative dissolution of pyrite relates to its environmental impact through ARD generation. Therefore, an understanding of the processes and mechanisms governing pyrite dissolution facilitates the predictive modelling of ARD, which in turn can be used to inform effective geotechnical designs and rehabilitation strategies. The controlled laboratory conditions allow measurement of the discrete isotope fractionations and therefore the effects of ARD-related processes on different isotope systems can be inferred with greater certainty. Interpretation of results in the field are therefore strengthened by laboratory experiments.
References
- Krouse, H.R.; Mayer, B. Sulphur and Oxygen Isotopes in Sulphate. In Environmental Tracers in Subsurface Hydrology; Cook, P., Herczeg, A., Eds.; Kluwer Academic Publishers, 2000; pp. 195–231
- Taylor, B.E.; Wheeler, M.C.; Nordstrom, D. K. Stable Isotope Geochemistry of Acid Mine Drainage: Experimental Oxidation of Pyrite. Geochim. Cosmochim. Acta 1984, 48 (12), 2669–2678.https://doi.org/10.1016/0016-7037(84)90315-6.
- Nakai, N.; Jensen, M.L. The Kinetic Isotope Effect in the Bacterial Reduction and Oxidation of Sulfur. Geochim. Cosmochim. Acta 1964, 28 (12), 1893–1912. https://doi.org/10.1016/0016-7037(64)90136-X
- McCready, R.G.L.; Krouse, H.R. Sulfur Isotope Fractionation during the Oxidation of Elemental Sulfur by Thiobacilli in a Solonetzic Soil. Can. J. Soil Sci. 1982, 62 (1), 105–110. https://doi.org/10.4141/cjss82-012.
- Balci, N.; Shanks, W.C.; Mayer, B.; Mandernack, K.W. Oxygen and Sulfur Isotope Systematics of Sulfate Produced by Bacterial and Abiotic Oxidation of Pyrite. Geochim. Cosmochim. Acta 2007, 71 (15), 3796–3811. https://doi.org/10.1016/j.gca.2007.04.017.
- Pisapia, C.; Chaussidon, M.; Mustin, C.; Humbert, B. O and S Isotopic Composition of Dissolved and Attached Oxidation Products of Pyrite by Acidithiobacillus Ferrooxidans: Comparison with Abiotic Oxidations. Geochim. Cosmochim. Acta 2007, 71 (10), 2474–2490.https://doi.org/10.1016/j.gca.2007.02.021.
- Heidel, C.; Tichomirowa, M. The Isotopic Composition of Sulfate from Anaerobic and Low Oxygen Pyrite Oxidation Experiments with Ferric Iron — New Insights into Oxidation Mechanisms. Chem. Geol. 2011, 281 (3–4), 305–316. https://doi.org/10.1016/j.chemgeo.2010.12.017.
- Brunner, B.; Yu, J.Y.; Mielke, R.E.; MacAskill, J.A.; Madzunkov, S.; McGenity, T.J.; Coleman, M. Different Isotope and Chemical Patterns of Pyrite Oxidation Related to Lag and Exponential Growth Phases of Acidithiobacillus Ferrooxidans Reveal a Microbial Growth Strategy. Earth Planet. Sci. Lett. 2008, 270 (1–2), 63–72. https://doi.org/10.1016/j.epsl.2008.03.019.
- Lefticariu, L.; Behum, P.; Bender, K.; Lefticariu, M. Sulfur Isotope Fractionation as an Indicator of Biogeochemical Processes in an AMD Passive Bioremediation System. Minerals 2017, 7 (3), 41. https://doi.org/10.3390/min7030041.
- Alvaro, A.; Roldán, F.V. Stable Isotope Composition of Acid Mine Drainage Minerals from San Miguel Massive Sulphide Mine Waste. In Securing the Future: Mining, Metals and the Environment in a Sustainable Society, Proceedings of the 8th International Conference on Acid Rock Drainage, Skellefteå, Sweden, 2009; 166–176.
- Gallo, A.A.; Roldán, F.V. Stable Isotope Study in Iron-Rich Sulphates from San Miguel Mine Wastes (Iberian Pyrite Belt, Spain). Rev. la Soc. Española Mineral. 2009, 11, 27–28.
- Schippers, A.; Sand, W. Bacterial Leaching of Metal Sulfides Proceeds by Two Indirect Mechanisms via Thiosulfate or via Polysulfides and Sulfur. Appl. Environ. Microbiol. 1999, 65 (1), 319–321.
- Rimstidt, D.D.; Vaughan, D.J. Pyrite Oxidation: A State-of-the-Art Assessment of the Reaction Mechanism. Geochim. Cosmochim. Acta 2003, 67 (5), 873–880. https://doi.org/10.1016/S0016-7037(02)01165-1
- Moses, C.O.; Herman, J.S. Pyrite Oxidation at Circumneutral pH. Geochim. Cosmochim. Acta 1991, 55 (2), 471–482. https://doi.org/10.1016/0016-7037(91)90005-P.
- Borda, M.J.; Strongin, D.R.; Schoonen, M.A.A. Vibrational Spectroscopic Study of Pyrite Oxidation. Am. Mineral. 2003, 88, 1318–1323.
- Descostes, M.; Vitorge, P.; Beaucaire, C. Pyrite Dissolution in Acidic Media. Geochim. Cosmochim. Acta 2004, 68 (22), 4559–4569. https://doi.org/10.1016/j.gca.2004.04.012.
- Goldhaber, M.B. Experimental Study of Metastable Sulfur Oxyanion Formation during Pyrite Oxidation at pH 6-9 and 30oC. Am. J. Sci. 1983, 283 (3), 193–217. https://doi.org/10.2475/ajs.283.3.193.
- Schippers, A.; Jozsa, P.G.; Sand, W. Sulfur Chemistry in the Leaching of Pyrite. Appl. Environm. Microbiol. 1996, 62 (9), 3424–3431. https://doi.org/10.3354/aei00166.
- Toran, L.; Harris, R.F. Interpretation of Sulfur and Oxygen Isotopes in Biological and Abiological Sulfide Oxidation. Geochim. Cosmochim. Acta 1989, 53 (9), 2341–2348. https://doi.org/10.1016/0016-7037(89)90356-6.
- McKibben, M.A.; Barnes, H.L. Oxidation of Pyrite in Low Temperature Acidic Solutions: Rate Laws and Surface Textures. Geochim. Cosmochim. Acta 1986, 50 (7), 1509–1520. https://doi.org/10.1016/0016-7037(86)90325-X.
- Luther, G.W. Pyrite Oxidation and Reduction: Molecular Orbital Theory Considerations. Geochim. Cosmochim. Acta 1987, 51 (12), 3193–3199. https://doi.org/10.1016/0016-7037(87)90127-X.
- Moses, C.O.; Nordstrom, K.D.; Herman, J.S.; Mills, A.L. Aqueous Pyrite Oxidation by Dissolved Oxygen and by Ferric Iron. Geochim. Cosmochim. Acta 1987, 51 (6), 1561–1571. https://doi.org/10.1016/0016-7037(87)90337-1.
- Demoisson, F.; Mullet, M.; Humbert, B. Pyrite Oxidation in Acidic Medium: Overall Reaction Pathway. Surf. Interface Anal. 2008, 40 (3–4), 343–348. https://doi.org/10.1002/sia.2720.
- Edraki, M.; Golding, S.D.; Baublys, K.A.; Lawrence, M.G. Hydrochemistry, Mineralogy and Sulfur Isotope Geochemistry of Acid Mine Drainage at the Mt. Morgan Mine Environment, Queensland, Australia. Appl. Geochemistry 2005, 20 (4), 789–805.https://doi.org/10.1016/j.apgeochem.2004.11.004.
- Dole, M.; Lane, G.A.; Rudd, D.P.; Zaukelies, D.A. Isotopic Composition of Atmospheric Oxygen and Nitrogen. Geochim. Cosmochim. Acta 1954, 6 (2–3), 65–78.
- Epstein, S.; Mayeda, T. Variations in the 18O Content of Waters from Natural Sources. Geochim. Cosmochim. Acta 1953, 4, 213–224.
- Taylor, B.E.; Wheeler, M.C. Sulfur- and Oxygen-isotope Geochemistry of Acid Mine Drainage in the Western United States. In Environmental Geochemistry of Sulfide Oxidation; Alpers, C.N., Blowes, D.W., Eds.; American Chemical Society Symposium Series: Washington DC, USA, 1993; Volume 550, pp. 481–514.
- Van Stempvoort, D.R.; Krouse, H.R. Controls of δ18O in Sulfate: Review of Experimental Data and Application to Specific Environments. In Environmental Geochemistry of Sulfide Oxidation; Alpers, C.N., Blowes, D.W., Eds.; American Chemical Society Symposium Series: Washington DC, USA, 1993; Volume 550, pp. 446–480.
- Johnson, C.M.; Skulan, J.L.; Beard, B.L.; Sun, H.; Nealson, K.H.; Braterman, P.S. Isotopic fractionation between Fe(III) and Fe(II) in aqueous solutions. Earth. Planet. Sci. Lett. 2002, 195 (1–2), 141–153. https://doi.org/10.1016/S0012-821X(01)00581-7.
- Anbar, A.D.; Jarzecki, A.A.; Spiro, T.G. Theoretical investigation of iron isotope fractionation between Fe(H2O)63+ and Fe(H2O)62+: Implications for iron stable isotope geochemistry. Geochim. Cosmochim. Acta. 2005, 69 (4), 825–837. https://doi.org/10.1016/j.gca.2004.06.012
- Skulan, J.L.; Beard, B.L.; Johnson, C.M. Kinetic and equilibrium Fe isotope fractionation between aqueous Fe(III) and hematite. Geochim. Cosmochim. Acta 2002, 66 (17), 2995–3015.https://doi.org/10.1016/S0016-7037(02)00902-X
- Fernandez, A.; Borrok, D.M. Fractionation of Cu, Fe, and Zn Isotopes during the Oxidative Weathering of Sulfide-Rich Rocks. Chem. Geol. 2009, 264 (1–4), 1–12.https://doi.org/10.1016/j.chemgeo.2009.01.024
- Nordstrom, K.D; Southam, G. Geomicrobiology of Sulfide Mineral Oxidation. In Geomicrobiology: Interactions between Microbes and Minerals: reviews in mineralogy; Banfield, J.F., Nealson, K.H., Eds.; Mineralogical Society of America: Washington DC, 1997; Volume 35, pp. 361–390.
- Croal, L.R.; Johnson, C.M.; Beard, B.L.; Newman, D.K. Iron isotope fractionation by Fe(II)-oxidizing photoautotrophic bacteria1. Geochim. Cosmochim. Acta 2004, 68 (6), 1227–42.https://doi.org/10.1016/j.gca.2003.09.011
- Balci, N.; Bullen, T. D.; Witte-Lien, K.; Shanks, W. C.; Motelica, M.; Mandernack, K. W. Iron Isotope Fractionation during Microbially Stimulated Fe(II) Oxidation and Fe(III) Precipitation. Geochim. Cosmochim. Acta 2006, 70 (3), 622–639. https://doi.org/10.1016/j.gca.2005.09.025
- Pérez Rodríguez, N.; Khoshkhoo, M.; Sandström, Å.; Rodushkin, I.; Alakangas, L.; Öhlander, B. Isotopic Signature of Cu and Fe during Bioleaching and Electrochemical Leaching of a Chalcopyrite Concentrate. Int. J. Miner. Process. 2015, 134, 58–65.https://doi.org/10.1016/j.minpro.2014.11.010.

