 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Veronika. Sexl | + 1789 word(s) | 1789 | 2020-12-03 09:22:56 |
Video Upload Options
Despite the development of targeted therapies and novel inhibitors, cancer remains an undefeated disease. Resistance mechanisms arise quickly and alternative treatment options are urgently required. Protein kinases as signaling switchboards are frequently deregulated in cancer and signify vulnerable nodes and potential therapeutic targets.
1. Introduction
Tumorigenesis requires that cells hijack regulatory networks to obtain their full oncogenic potential. The MAPK (mitogen-activated protein kinase) pathway is a tightly regulated signaling cascade frequently mutated or deregulated in cancer. Extra- and intracellular stimuli activate the MAPK cascade that regulates a broad variety of cellular programs including proliferation, differentiation, stress responses and apoptosis [1]. The best-studied MAPK pathways are the extracellular signal-regulated kinase (ERK) and stress-activated MAPK (c-Jun N-terminal kinase JNK and p38) pathways (Figure 1). While the ERK pathway is predominantly activated by growth factors to drive survival and cell growth, activation of the JNK or p38 pathway is accomplished by environmental stressors and inflammatory cytokines and is associated with apoptosis and growth inhibition [1]. MAPK cascades are structured in three core protein kinases (MAP3K, MAP2K and MAPK), which allows a tight regulation [1] (Figure 1).
The MAPK pathway interferes with the cell cycle machinery at many stages [1][2]. Transcriptional regulation of cell cycle components by MAPKs links cyclin-dependent kinases (CDKs) to the MAP kinase cascade. MAPKs regulate the expression levels of D-type cyclins, key players of the cell cycle [2]. While ERK signaling stimulates cyclin D1 expression upon growth factor exposure, p38 signaling suppresses cyclin D1 expression [2]. Changing the levels of D-type cyclins impacts on the activation of their cell cycle partners, the cyclin-dependent kinases 4 and 6 (CDK4 and CDK6). CDK4 and its close homologue CDK6 play a crucial role in various cancer types. Both are part of the G1 cell cycle checkpoint and allow the cell to enter into the S-phase [3]. When bound to D-type cyclins, CDK4/6 phosphorylate the retinoblastoma (Rb) proteins to release Rb-mediated repression on E2F target genes that drive G1 to S progression [4][5] (Figure 1).
Another layer of complexity is added as the ERK pathway regulates cell cycle progression dependent on the intensity and duration of the stimulating signal [6][7]. Whereas pronounced Raf activation triggers the induction of CDK4/6 inhibitor p21Cip1 and inhibits G1/S progression, moderate Raf signaling promotes DNA synthesis and expression of cyclin D1 [7].
In addition, p38 stabilizes the p21Cip1 protein [8]. High p21Cip1 levels induce a G1 arrest by binding to and inactivating G1 phase CDKs [9]. However, p38 also affects later phases of the cell cycle by interfering at the G2/M checkpoint. p38 activation initiates a G2 arrest as the immediate response to UV exposure [10][11]. Thus p38 serves as a sensor of DNA damage and puts the break on the cell cycle at the G2/M border.
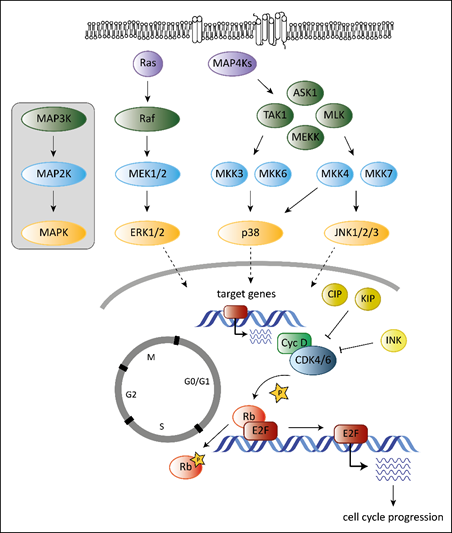
Figure 1. Overview of the best-studied MAPK signaling cascades and their interplay with the cell cycle machinery. MAPK cascades consist of three core kinases. The general pattern of the core kinases is shown in the box on the left. The three core kinases are activated by upstream kinases (MAP4Ks, Ras). Activation is indicated by the arrows. Activated MAPKs translocate into the nucleus (indicated by the dashed arrows) and induce expression of their target genes. Target genes of MAPKs are among others genes encoding D-type cyclins. They bind and activate cell cycle kinases CDK4 and 6 leading to phosphorylation on Rb (indicated by the star symbol) and to release of Rb-mediated repression of E2F target genes resulting in progression of the cell cycle to the S-phase. Members of the CIP/KIP and the INK protein families inhibit CDK4/6 as indicated by the “T-lines”.
The MAPK cascade and the CDK4/6 signaling complex are frequently deregulated or mutated in a variety of cancer types, which prompted the development of small molecular weight inhibitors that made it to clinic. Their impact is limited by resistance development, which evolves rapidly when drugs are used as monotherapy. One important strategy to avoid resistance development is the use of drug combinations. On the one hand combinational treatment may lead to a more efficient eradication of tumor cells if the drugs act synergistically. On the other hand it may support resensitization of tumor cells with acquired resistance. Tumors treated with MAPK inhibitors often acquire resistance after only a short period. One prominent mechanism of acquired resistance in this context is reactivation of the MAPK pathway and upregulation of cyclin D1 [12]. Simultaneous inhibition of CDK4/6, the main binding partner of cyclin D thus represents a promising treatment strategy to block proliferation of tumor cells and delay or overcome resistance to MAPK inhibitors.
We here discuss the interference of CDK6 and MAPK cascades in cancer.
2. The Role of CDK6 in Cancer
Cyclin D–CDK4/6 activation is frequently deregulated in cancer. Despite overlapping functions in cell cycle regulation, CDK4 and CDK6 dysregulation is linked to different cancer types. High levels of CDK4 are common in sarcoma [13], glioma [14] and melanoma [15]. While a mutant form of CDK4 associated with insensitivity towards the inhibitory INK4 proteins is found in familiar melanoma [15], no mutations have been identified so far in CDK6. Alterations for CDK6 include overexpression, which is frequently found in hematologic malignancies [16][17]. The unique functions of CDK6 as a transcriptional regulator in the hematopoietic system, which is not shared by CDK4 may partly explain this pattern.
CDK6 may exert tumor promoting and tumor suppressing functions. On the one hand, the kinase-dependent function of CDK6 in the cell cycle machinery is exploited by rapidly proliferating tumor cells to rush through the cell cycle. In addition, the function of CDK6 as transcriptional regulator interferes with tumor progression at multiple levels. As transcriptional regulator, CDK6 together with the activator protein 1 (AP-1) member c-JUN, controls vascular endothelial growth factor A (VEGF-A) expression to regulate angiogenesis [18]. AP-1 is a dimeric complex composed of members of the JUN, FOS, ATF and MAF protein families. Different AP-1 dimers bind distinct DNA sites to regulate transcription of their target genes [19]. The proto-oncogene c-Jun not only drives angiogenesis together with CDK6 but may also regulate the expression of CDK6 itself. C-Jun binds to the CDK6 promoter region and thereby prevents promoter methylation and thus silencing to maintain high CDK6 expression levels. The strong interplay of CDK6 and c-Jun leads to the suggestion that CDK6 might also interact with other AP-1 members to fulfill specific functions. This relationship needs to be analyzed in great detail for different disease entities to understand and develop novel treatment options.
At the flip side of the coin, CDK6 suppresses tumorigenesis by forming complexes with STAT3 and cyclin D to induce expression of the tumor suppressor p16INK4a [18][20]. Thereby, CDK6 induces its own inhibitor p16INK4a, which is viewed as a feedback loop and internal control mechanism protecting the cell from high CDK6 levels and uncontrolled cell proliferation. Thus, the presence of p16INK4a determines whether CDK6 acts as a tumor-suppressor. In the absence of p16INK4A CDK6 turns into a proto-oncogene [18][20]. In addition, the balanced expression of CDK4 and CDK6 is crucial to allow CDK6 to act as transcriptional regulator as has been shown in melanoma cells [21]. Only the presence of CDK4, which takes over cell cycle regulation, frees CDK6 to act as a transcriptional regulator [21].
A high expression of Cyclin D1 and CDK4 is frequently found in breast cancer and deregulation of this pathway contributes to uncontrolled proliferation in hormone receptor-positive breast cancer [22][23][24][25]. Estradiol allows for the rapid progression through the G1 phase of the cell cycle by mediating activation of G1 CDKs, enhancing expression of cyclin D1 and inactivation of the CDK inhibitor p27KIP [26]. In prostate cancer, androgen receptor-dependent transcription is stimulated by direct binding of CDK6 [27]. Overexpression of CDK6 thereby contributes to prostate cancer progression.
The frequent overexpression of CDK6 in leukemia and lymphoma has put a focus on CDK6 dependent functions in the hematopoietic system. CDK6 binds RUNX1, a master regulator of hematopoietic differentiation and blocks RUNX1 binding to DNA and thereby myeloid differentiation [28]. Interference with myeloid differentiation was also observed in MLL/AF9-driven myeloid leukemia, where CDK6 inhibition triggered differentiation [29].
3. The Role of MAPKs in Cancer
MAPKs are activated by various stimuli and are involved in growth control; it is thus not surprising that dysregulation is linked to initiation and maintenance of numerous cancer types. The Ras/Raf/MEK/ERK signaling cascade is predominantly activated by growth factors and promotes cell proliferation and survival [30][31]. As such, it represents one of the most frequently mutated pathways in cancer [30]. Mutations occur in different components of the pathway with RAS and RAF being mutational hotspots and considered oncogenic drivers. They are most commonly mutated in lung, colon, pancreatic cancer and melanoma [30][32]. Mutations keep the Ras family of GTPases in an active GTP bound state [32] and predominantly affect KRAS [33]. Mutations in BRAF are the most common RAF mutations with one specific point mutation (BRAFV600E) being present in more than 90% of cases [33][34]. Mutations of MEK and ERK are less frequent, but are associated with a poor prognosis in many cancer types and do not co-occur with RAS or RAF mutations [33].
Aberrant ERK signaling is also described in hematopoietic malignancies and promotes cell growth and survival [35]. A large subset of primary AML shows constitutively activated ERK signaling indicative for a crucial role in leukemogenesis [36]. In chronic myeloid leukemia (CML), the oncogenic fusion kinase BCR-ABL activates MAPK pathways that are suggested to promote leukemogenesis [32]. BCR-ABL inhibition is synergistic with inhibition of the ERK pathway and induces apoptosis in BCR-ABL positive cells [37].
Deregulated ERK signaling activates transcription factors that contribute to transformation. Transcription and activation of proto-oncogenic members of the AP-1 family depends on MAPK signaling. Mutations in the MAPK pathway affect AP-1 functions, which contributes to transformation. ERK, p38 and JNK regulate the expression of different AP-1 members via ternary complex factors (TCFs; Figure 2). AP-1 members are also subject to phosphorylation by the MAPK pathway as has been shown for the c-FOS protein [38]. Another example is the activating transcription factor 2 (ATF2), which is a direct target of p38 [39]. Phosphorylated ATF2 forms heterodimers with members of the Jun family to associate with AP-1 binding sites, further enhancing expression of c-Jun [40].
Members of the AP-1 family interact with CDK6 on chromatin to regulate transcription [18]. AP-1 transcription factors might thereby represent the link between the MAPK pathway and CDK6 to enable the crosstalk between these programs contributing to transformation and tumor maintenance.
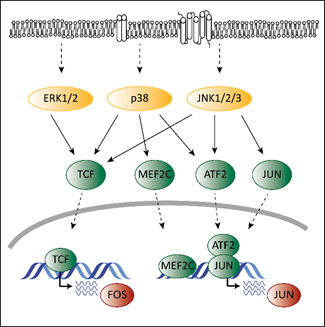
Figure 2. The interplay of CDK4/6 and the MAPK pathways in the context of AP-1. Adapted from [19][41]. Activating signals pass through the MAPK cascade until they reach MAPKs (indicated by the dashed arrows at the top). MAPKs activate ternary complex factors (TCF), MEF2C, ATF2 and JUN (indicated by the arrows) that translocate into the nucleus (indicated by the lower dashed arrows) and induce expression of FOS and JUN.
References
- M Raman; Weiyi Chen; Melanie H. Cobb; Differential regulation and properties of MAPKs. Oncogene 2007, 26, 3100-3112, 10.1038/sj.onc.1210392.
- Josée N. Lavoie; Gilles L'allemain; Anne Brunet; Rolf Müller; Jacques Pouysségur; Cyclin D1 Expression Is Regulated Positively by the p42/p44MAPKand Negatively by the p38/HOGMAPKPathway. Journal of Biological Chemistry 1996, 271, 20608-20616, 10.1074/jbc.271.34.20608.
- M Meyerson; E Harlow; Identification of G1 kinase activity for cdk6, a novel cyclin D partner.. Molecular and Cellular Biology 1994, 14, 2077-2086, 10.1128/mcb.14.3.2077.
- A-S Tigan; Florian Bellutti; Karoline Kollmann; Graham Tebb; V Sexl; CDK6—a review of the past and a glimpse into the future: from cell-cycle control to transcriptional regulation. Oncogene 2015, 35, 3083-3091, 10.1038/onc.2015.407.
- Marie Classon; Ed Harlow; The retinoblastoma tumour suppressor in development and cancer. Nature Reviews Cancer 2002, 2, 910-917, 10.1038/nrc950.
- Kristin Roovers; Richard K. Assoian; Integrating the MAP kinase signal into the G1 phase cell cycle machinery. BioEssays 2000, 22, 818-826, 10.1002/1521-1878(200009)22:9<818::aid-bies7>3.0.co;2-6.
- A Sewing; B Wiseman; A C Lloyd; H Land; High-intensity Raf signal causes cell cycle arrest mediated by p21Cip1.. Molecular and Cellular Biology 1997, 17, 5588-5597, 10.1128/mcb.17.9.5588.
- Geum-Yi Kim; Stephen E. Mercer; Daina Z. Ewton; Zhongfa Yan; Kideok Jin; Eileen Friedman; The Stress-activated Protein Kinases p38α and JNK1 Stabilize p21Cip1 by Phosphorylation. Journal of Biological Chemistry 2002, 277, 29792-29802, 10.1074/jbc.m201299200.
- Zhongfa Yan; Geum-Yi Kim; Xiaobing Deng; Eileen Friedman; Transforming Growth Factor β1 Induces Proliferation in Colon Carcinoma Cells by Ras-dependent, smad-independent Down-regulation of p21cip1. Journal of Biological Chemistry 2002, 277, 9870-9879, 10.1074/jbc.m107646200.
- Dmitry V. Bulavin; Yuichiro Higashimoto; Ian J. Popoff; William A. Gaarde; Venkatesha Basrur; Olga Potapova; Ettore Appella; Albert J. Fornace Jr; Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature 2001, 411, 102-107, 10.1038/35075107.
- Anupama Munshi; Rajagopal Ramesh; Mitogen-Activated Protein Kinases and Their Role in Radiation Response. Genes & Cancer 2013, 4, 401-408, 10.1177/1947601913485414.
- José Luís Manzano; Laura Layos; Cristina Bugés; María De Los Llanos Gil; Laia Vila; Eva Martínez-Balibrea; Anna Martínez-Cardús; Resistant mechanisms to BRAF inhibitors in melanoma. Annals of Translational Medicine 2016, 4, 237-237, 10.21037/atm.2016.06.07.
- Z A Khatib; H Matsushime; M Valentine; D N Shapiro; C J Sherr; A T Look; Coamplification of the CDK4 gene with MDM2 and GLI in human sarcomas.. Cancer Research 1993, 53, 5535–5541.
- E E Schmidt; Koichi Ichimura; G Reifenberger; V P Collins; CDKN2 (p16/MTS1) gene deletion or CDK4 amplification occurs in the majority of glioblastomas.. Cancer Research 1994, 54, 6321–6324.
- Thomas Wolfel; M. Hauer; Jenny Schneider; Myrna G Serrano; Catherine Wolfel; E. Klehmann-Hieb; Etienne De Plaen; Thomas Hankeln; K H Meyer Zum Büschenfelde; Dale L Beach; et al. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science 1995, 269, 1281-1284, 10.1126/science.7652577.
- M. Chilosi; C. Doglioni; Z. Yan; M. Lestani; F. Menestrina; C. Sorio; A. Benedetti; F. Vinante; G. Pizzolo; G. Inghirami; et al. Differential expression of cyclin-dependent kinase 6 in cortical thymocytes and T-cell lymphoblastic lymphoma/leukemia.. The American Journal of Pathology 1998, 152, 209-217.
- Stefan Nagel; Ellen Leich; Hilmar Quentmeier; Charles R Meyer; Manfred Kaufmann; Hans G Drexler; Andreas Zettl; Andreas Rosenwald; Roderick A F MacLeod; Amplification at 7q22 targets cyclin-dependent kinase 6 in T-cell lymphoma. Leukemia 2007, 22, 387-392, 10.1038/sj.leu.2405028.
- Karoline Kollmann; Gerwin Heller; Christine Schneckenleithner; Wolfgang Warsch; Ruth Scheicher; Rene G. Ott; Markus Schäfer; Sabine Fajmann; Michaela Schlederer; Ana-Iris Schiefer; et al.Ursula ReichartMatthias MayerhoferChristoph HoellerSabine Zöchbauer-MüllerDontscho KerjaschkiChristoph BockLukas KennerGerald HoeflerMichael FreissmuthAnthony R. GreenRichard MorigglMeinrad BusslingerMarcos MalumbresVeronika Sexl A Kinase-Independent Function of CDK6 Links the Cell Cycle to Tumor Angiogenesis. Cancer Cell 2013, 24, 167-181, 10.1016/j.ccr.2013.07.012.
- Robert Eferl; Erwin F. Wagner; AP-1: a double-edged sword in tumorigenesis. Nature Reviews Cancer 2003, 3, 859-868, 10.1038/nrc1209.
- Karoline Kollmann; Veronika Sexl; CDK6 and p16INK4A in lymphoid malignancies. Oncotarget 2013, 4, 1858-1859, 10.18632/oncotarget.1541.
- Karoline Kollmann; Coralie Briand; Florian Bellutti; Nikolaus Schicher; Stefan Blunder; Markus Zojer; Christoph Hoeller; The interplay of CDK4 and CDK6 in melanoma. Oncotarget 2019, 10, 1346-1359, 10.18632/oncotarget.26515.
- Qunyan Yu; Ewa Sicinska; Yan Geng; Marie Ahnström; Agnieszka Zagozdzon; Yinxin Kong; Humphrey Gardner; Hiroaki Kiyokawa; Lyndsay N. Harris; Olle Stål; et al.Piotr Sicinski Requirement for CDK4 kinase function in breast cancer. Cancer Cell 2006, 9, 23-32, 10.1016/j.ccr.2005.12.012.
- Jirina Bartkova; Jiri Lukas; Heiko Müller; Dorrit Lützhøt; Michael Strauss; Jiri Bartek; Cyclin D1 protein expression and function in human breast cancer. International Journal of Cancer 1994, 57, 353-361, 10.1002/ijc.2910570311.
- C. Dickson; V. Fantl; C. Gillett; S. Brookes; J. Bartek; R. Smith; C. Fisher; D. Barnes; G. Peters; Amplification of chromosome band 11q13 and a role for cyclin D1 in human breast cancer. Cancer Letters 1995, 90, 43-50, 10.1016/0304-3835(94)03676-a.
- Ben O'leary; Richard S. Finn; Ben O'Leary Nicholas C. Turner; Treating cancer with selective CDK4/6 inhibitors. Nature Reviews Clinical Oncology 2016, 13, 417-430, 10.1038/nrclinonc.2016.26.
- J. S. Foster; Estrogen regulates activity of cyclin-dependent kinases and retinoblastoma protein phosphorylation in breast cancer cells. Molecular Endocrinology 1996, 10, 488-498, 10.1210/me.10.5.488.
- Jin T. E. Lim; Mahesh Mansukhani; I Bernard Weinstein; Cyclin-dependent kinase 6 associates with the androgen receptor and enhances its transcriptional activity in prostate cancer cells. Proceedings of the National Academy of Sciences 2005, 102, 5156-5161, 10.1073/pnas.0501203102.
- T Fujimoto; K Anderson; Sten Eirik W. Jacobsen; S-I Nishikawa; Claus Nerlov; Cdk6 blocks myeloid differentiation by interfering with Runx1 DNA binding and Runx1-C/EBPα interaction. The EMBO Journal 2007, 26, 2361-2370, 10.1038/sj.emboj.7601675.
- Theresa Placke; Katrin Faber; Atsushi Nonami; Sarah L. Putwain; Helmut R. Salih; Florian H. Heidel; Alwin Krämer; David E. Root; David A. Barbie; Andrei V. Krivtsov; et al.Scott A. ArmstrongWilliam C. HahnBrian J. HuntlyStephen M. SykesMichael D. MilsomClaudia SchollStefan Fröhling Requirement for CDK6 in MLL-rearranged acute myeloid leukemia. Blood 2014, 124, 13-23, 10.1182/blood-2014-02-558114.
- Judith S. Sebolt-Leopold; Roman Herrera; Targeting the mitogen-activated protein kinase cascade to treat cancer. Nature Reviews Cancer 2004, 4, 937-947, 10.1038/nrc1503.
- P J Roberts; Channing J. Der; Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291-3310, 10.1038/sj.onc.1210422.
- Julian Downward; Targeting RAS signalling pathways in cancer therapy. Nature Reviews Cancer 2003, 3, 11-22, 10.1038/nrc969.
- Ufuk Degirmenci; Mei Wang; Jiancheng Hu; Targeting Aberrant RAS/RAF/MEK/ERK Signaling for Cancer Therapy. Cells 2020, 9, 198, 10.3390/cells9010198.
- Helen N Davies; Graham R. Bignell; Charles E Cox; Philip J Stephens; Sarah Edkins; Sheila Clegg; Jon W Teague; Hayley Woffendin; Mathew J. Garnett; William Bottomley; et al.Neil DavisEd DicksRebecca EwingYvonne FloydKristian GraySarah HallRachel HawesJaime HughesVivian KosmidouAndrew MenziesCatherine MouldAdrian G ParkerClaire StevensStephen WattSteven HooperRebecca M WilsonHiran JayatilakeBarry A. GustersonColin S CooperJanet ShipleyDarren HargraveKatherine Pritchard-JonesNorman J MaitlandGeorgia Chenevix-TrenchGregory J. RigginsDarell D. BignerGiuseppe PalmieriAntonio CossuAdrienne M FlanaganAndrew C NicholsonJudy W. C. HoSuet Y. LeungSiu T. YuenBarbara L. WeberHilliard F. SeiglerTimothy L. DarrowHugh PatersonRichard MaraisChristopher J. MarshallRichard WoosterMichael R. StrattonP. Andrew Futreal Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949-954, 10.1038/nature00766.
- Leonidas C. Platanias; Map kinase signaling pathways and hematologic malignancies. Blood 2003, 101, 4667-4679, 10.1182/blood-2002-12-3647.
- M Towatari; H Iida; M Tanimoto; H Iwata; M Hamaguchi; H Saito; Constitutive activation of mitogen-activated protein kinase pathway in acute leukemia cells. Leukemia 1997, 11, 479-484, 10.1038/sj.leu.2400617.
- Chunrong Yu; Geoffrey Krystal; Lyuba Varticovksi; Robert McKinstry; Mohamed Rahmani; Paul Dent; Steven Grant; Pharmacologic mitogen-activated protein/extracellular signal-regulated kinase kinase/mitogen-activated protein kinase inhibitors interact synergistically with STI571 to induce apoptosis in Bcr/Abl-expressing human leukemia cells.. Cancer Research 2002, 62, 188–199.
- A. J. Whitmarsh; R. J. Davis; Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. Journal of Molecular Medicine 1996, 74, 589-607, 10.1007/s001090050063.
- Joël Raingeaud; Shashi Gupta; Jeffrey Scott Rogers; Martin Dickens; Jiahuai Han; Richard J. Ulevitch; Roger J. Davis; Pro-inflammatory Cytokines and Environmental Stress Cause p38 Mitogen-activated Protein Kinase Activation by Dual Phosphorylation on Tyrosine and Threonine. Journal of Biological Chemistry 1995, 270, 7420-7426, 10.1074/jbc.270.13.7420.
- H Van Dam; D Wilhelm; I Herr; A Steffen; P Herrlich; P Angel; ATF-2 is preferentially activated by stress-activated protein kinases to mediate c-jun induction in response to genotoxic agents.. The EMBO Journal 1995, 14, 1798-1811.
- Karoline Kollmann; Gerwin Heller; Veronika Sexl; c-JUN prevents methylation of p16INK4a (and Cdk6): the villain turned bodyguard. Oncotarget 2011, 2, 422-427, 10.18632/oncotarget.279.


