Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Zheng, Y.; Xu, X.; Chi, F.; Cong, N. Pyroptosis. Encyclopedia. Available online: https://encyclopedia.pub/entry/35322 (accessed on 30 July 2026).
Zheng Y, Xu X, Chi F, Cong N. Pyroptosis. Encyclopedia. Available at: https://encyclopedia.pub/entry/35322. Accessed July 30, 2026.
Zheng, Yu, Xinda Xu, Fanglu Chi, Ning Cong. "Pyroptosis" Encyclopedia, https://encyclopedia.pub/entry/35322 (accessed July 30, 2026).
Zheng, Y., Xu, X., Chi, F., & Cong, N. (2022, November 19). Pyroptosis. In Encyclopedia. https://encyclopedia.pub/entry/35322
Zheng, Yu, et al. "Pyroptosis." Encyclopedia. Web. 19 November, 2022.
Copy Citation
Ischemia-reperfusion (I/R) injury, uncommon among patients suffering from myocardial infarction, stroke, or acute kidney injury, can result in cell death and organ dysfunction. Previous studies have shown that different types of cell death, including apoptosis, necrosis, and autophagy, can occur during I/R injury. Pyroptosis, which is characterized by cell membrane pore formation, pro-inflammatory cytokine release, and cell burst, and which differentiates itself from apoptosis and necroptosis, has been found to be closely related to I/R injury.
pyroptosis
ischemia-reperfusion injury
gasdermin
therapeutic target
1. Introduction
Ischemia-reperfusion (I/R) injury is caused by the resumption of blood supply (reperfusion) to an organ or tissue after a period of restricted blood supply, resulting in pathological processes that can lead to cell death and organ dysfunction. I/R injury can occur in many vital organs, including the brain, heart, and kidneys; it can also occur in less noticed organs, such as the cochlea and vestibule in the ear. The mechanisms of I/R injury comprise oxidative stress, sterile inflammation, calcium overload, mitochondrial dysfunction, and activation of various cell death pathways—including, but not limited to, apoptosis, necrosis, and autophagy [1]. Pyroptosis is another type of regulated cell death, characterized by cell membrane pore formation, pro-inflammatory cytokine release, and cell lysis [2]. It has previously been found that pyroptosis is closely related to the development of tumors [3]. Furthermore, recent studies have shown that pyroptosis also plays an important role in I/R injury. Because the current treatment and management of I/R injury are far from satisfactory [4], advances in understanding of the mechanisms of pyroptosis and its role in I/R injury may lead to innovative therapeutic strategies for treating patients with I/R-associated tissue inflammation and organ dysfunction.
2. Overview of Pyroptosis
Pyroptosis was first discovered by Zychlinsky et al. in 1992. They reported that the death of Shigella flexneri-infected macrophages was dependent on caspase-1 rather than caspase-3, which was then thought to be only involved in apoptosis [5]. In 1996, Chen et al. found that the invasion plasmid antigen B of Shigella flexneri could bind to the ICE (interleukin-1β-converting enzyme, caspase-1) directly, leading to the activation of enzymes in the infected macrophages and ultimately resulting in the death of those infected macrophages [6]. In 1999, Hersh et al. discovered that caspase-1 plays an indispensable role in this specific cell death mode, as caspase-1-knockout macrophages could not be induced into death by Shigella flexneri infection [7]. In 2001, this form of regulated cell death was named “pyroptosis” by Cookson and Brennan after they found a similar phenomenon in macrophages infected with Salmonella typhimurium [8]. Seven years later, fragmented DNA and a damaged cell membrane were found by Cookson et al. during the process of pyroptosis, which led to the release of intracellular content and triggered a severe inflammatory response [9]. In 2011, Kayagaki et al. found that caspase-11 was able to induce mouse macrophage death, which is essentially similar to caspase-1-mediated pyroptosis. They termed this caspase-11-mediataed pyroptosis as the “non-canonical pathway of pyroptosis” [10].
In 2015, Shao et al. conducted CRISPR-Cas9 nuclease screening for the whole genome of caspase-11- and caspase-1-mediated pyroptosis of mouse bone marrow macrophages, and they eventually identified gasdermin D (GSDMD) as the inflammatory caspase substrate through which caspases activated pyroptosis [11]. The study further proved that caspase-1 and caspase-4/5/11 specifically cleaved the linker between the amino-terminal gasdermin-N and the carboxy-terminal gasdermin-C domains in GSDMD, ultimately leading to cell pyroptosis, which was triggered only by the gasdermin-N domain.
GSDMD is a member of the human gasdermin superfamily, which consists of gasdermin A/B/C/D (GSDMA/B/C/D), gasdermin E (GSDME, also known as DFNA5), and DFNB59 (PJVK); the mouse gasdermin superfamily is composed of GSDMA1-3, GSDMC1-4, GSDMD, GSDME, and PJVK [12][13][14][15]. Among these proteins, GSDMD and GSDME are currently the most comprehensively researched in terms of pyroptotic death. Moreover, other than PJVK, these conserved proteins consist of two conserved domains, the N-terminal pore-forming domain (PFD) and the C-terminal repressor domain (RD) [16][17][18]. Most of the gasdermins’ N-terminals can trigger pyroptotic death, but this has not been found in PJVK so far [19][20]. In general, gasdermins maintain oligomerization through the interaction of two domains. When cells are stimulated by various factors, gasdermins are cleaved by certain caspases into the N-terminal PFD and the C-terminal RD. After dissociation, the PFD oligomerizes on the cell membrane to form pores and cause cell pyroptosis [21][22].
3. Mechanisms of Pyroptosis
It was formerly believed that there are two pathways of pyroptosis: one is the canonical pathway dependent on caspase-1, and the other is the non-canonical pathway dependent on human caspase-4/5 or mouse caspase-11. However, due to in-depth study in recent years, other pathways of pyroptosis have been found (Figure 1). The detailed mechanisms of pyroptosis and its pathways are discussed below.
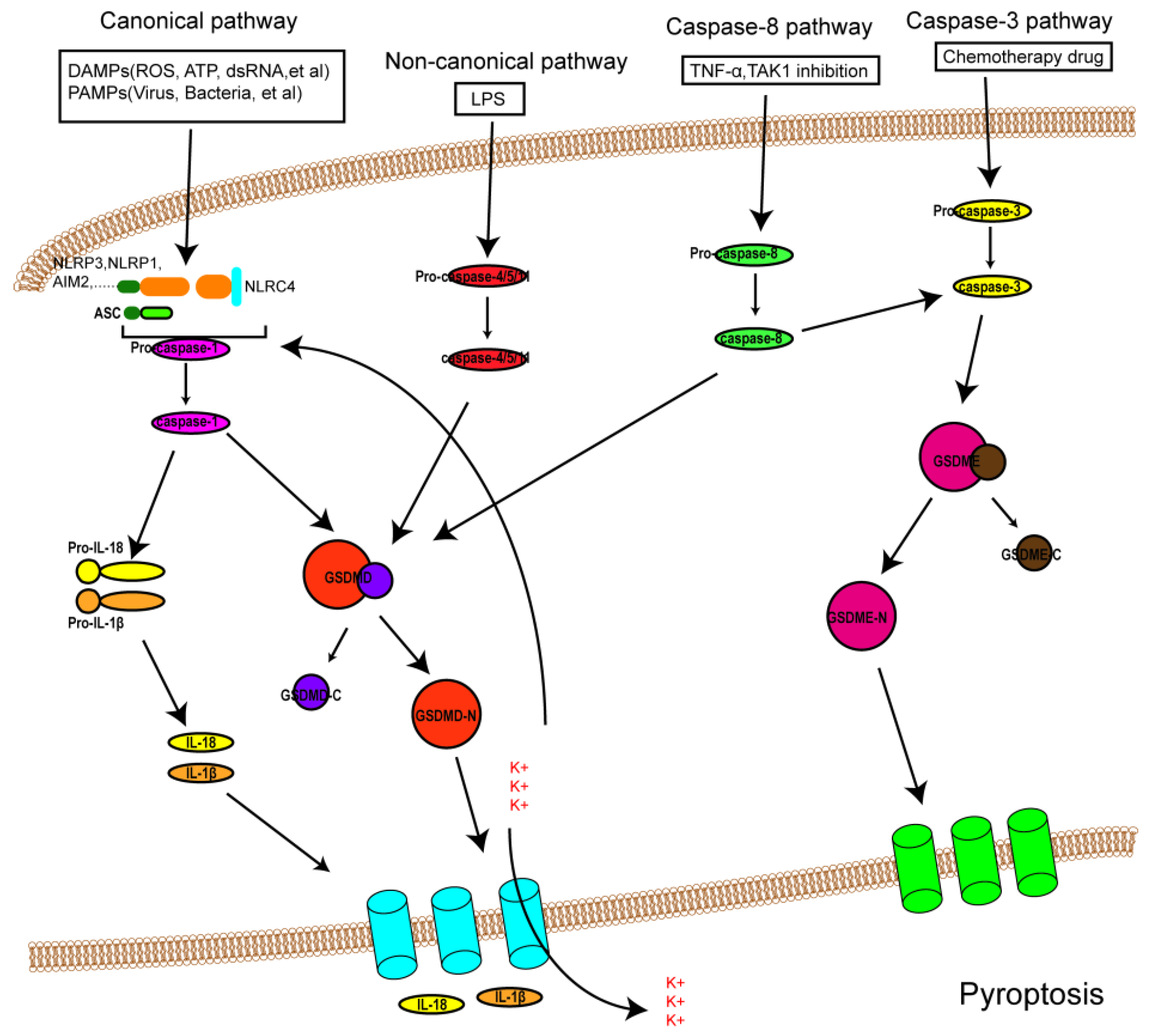
Figure 1. An overview of the mechanisms of pyroptosis. In the canonical pathway, PAMPs and DAMPs receive signaling molecule stimulation and assemble with pro-caspase-1 and ASC to form inflammasomes and active caspase-1. Activated caspase-1 cleaves GSDMD into GSDMD-N and GDSMD-C and lyses the precursors of IL-1β and IL-18 (pro-ILβ and pro-IL-18, respectively). GSDMD-N perforates the cell membrane by forming non-selective pores, further causing water influx and cell swelling and bursting. In addition, IL-1β and IL-18 are secreted from the pores formed by GSDMD-N. In the noncanonical pathway, LPS activates caspase-4/5 and caspase-11, triggering pyroptosis by cleaving GSDMD. In addition, the cleavage of GSDMD results in efflux of K+, ultimately giving rise to the assembly of an NLRP3 inflammasome. In the caspase-3-mediated pathway, active caspase-3 cleaves GSDME to form GSDME-N, inducing cell pyroptosis. In the caspase-8-mediated pathway, inhibiting TAK1 induces the activation of caspase-8, which cleaves GSDMD or GSDME and results in pyroptosis.
4. Pyroptosis and I/R Injury
I/R injury mainly revolves around various kinds of cell death and subsequent organ dysfunction. Previous studies have identified that necrosis, apoptosis, and autophagy-associated cell death are implicated in I/R injury [1]. Consequently, treatments targeted at these cell death patterns have also been investigated. Though some progress has been made in dealing with I/R injury so far, the outcomes are still far from satisfactory. Fortunately, the discovery of pyroptosis has brought new hope and impetus to the treatment of I/R injury, as an increasing number of studies have suggested that pyroptosis is involved in I/R injury (the link between pyroptosis and I/R injury was shown in Figure 2). This could allow the adoption of potentially feasible and effective approaches to alleviating I/R injury by targeting pyroptotic cell death. This section discusses the role of pyroptosis in I/R injury in different organs and possible therapeutic strategies to attenuate the intensity of I/R injury.
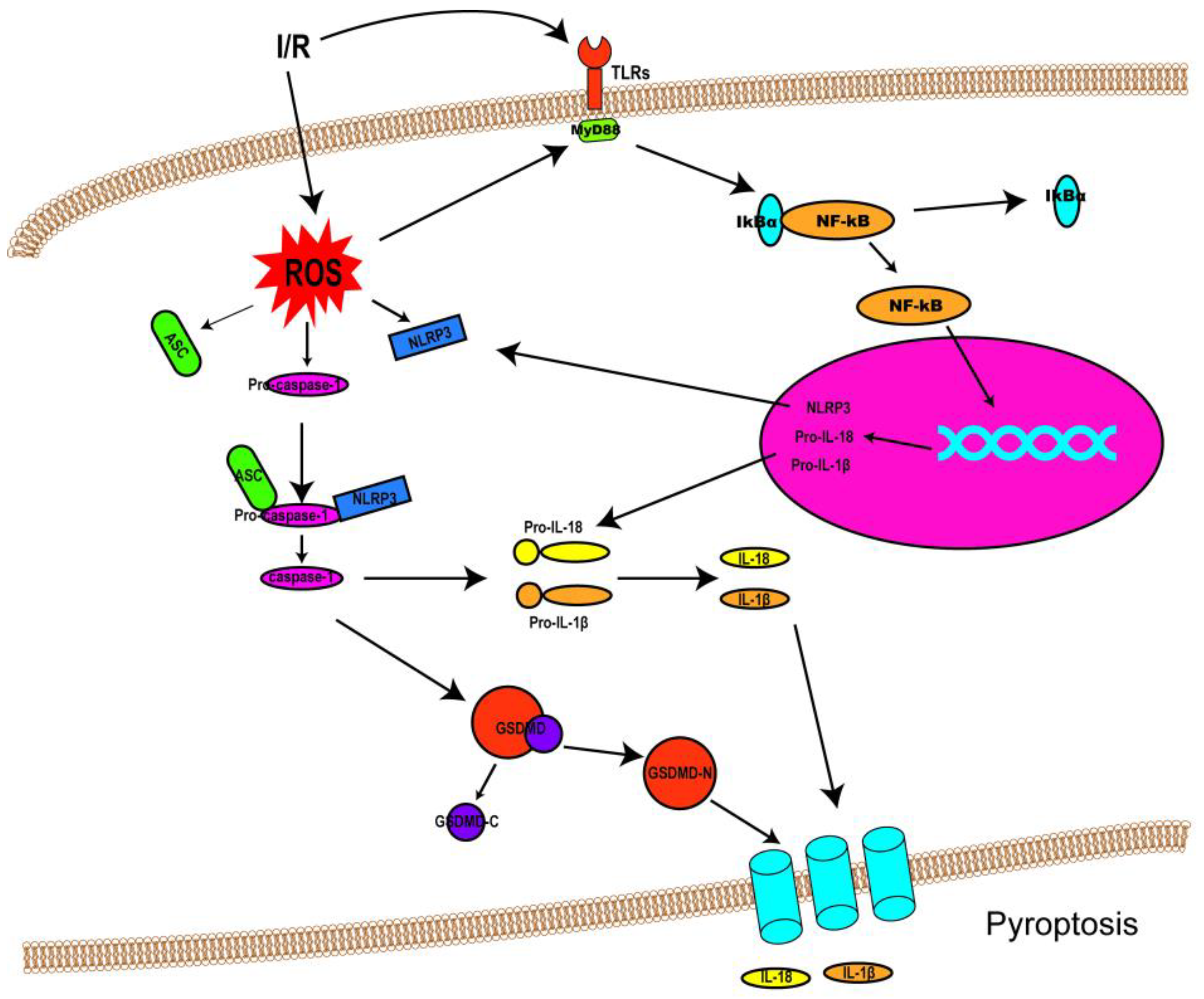
Figure 2. Schematic diagram demonstrating the link between pyroptosis and I/R injury. Following I/R insults, NF-κB signaling (priming) is activated by Toll-like receptors (TLRs), and ROS are also produced, resulting in NLRP3 inflammasome assembly and caspase-1 activation. Activated caspase-1 cleaves GSDMD into GSDMD-N and GDSMD-C and lyses the precursors of IL-1β and IL-18 (pro-ILβ and pro-IL-18, respectively) to form mature IL-1β and IL-18. Then, GSDMD-N oligomerizes and penetrates the cell membrane, forming non-selective pores on the membrane to release IL-1β and IL-18.
References
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion—From mechanism to translation. Nat. Med. 2011, 17, 1391–1401.
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022.
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and diseases. Signal Transduct. Target. Ther. 2021, 6, 128.
- Pantazi, E.; Bejaoui, M.; Folch-Puy, E.; Adam, R.; Rosello-Catafau, J. Advances in treatment strategies for ischemia reperfusion injury. Expert Opin. Pharmacother. 2016, 17, 169–179.
- Zychlinsky, A.; Prevost, M.C.; Sansonetti, P.J. Shigella flexneri induces apoptosis in infected macrophages. Nature 1992, 358, 167–169.
- Chen, Y.; Smith, M.R.; Thirumalai, K.; Zychlinsky, A. A bacterial invasin induces macrophage apoptosis by binding directly to ICE. EMBO J. 1996, 15, 3853–3860.
- Hersh, D.; Monack, D.M.; Smith, M.R.; Ghori, N.; Falkow, S.; Zychlinsky, A. The Salmonella invasin SipB induces macrophage apoptosis by binding to caspase-1. Proc. Natl. Acad. Sci. USA 1999, 96, 2396–2401.
- Brennan, M.A.; Cookson, B.T. Salmonella induces macrophage death by caspase-1-dependent necrosis. Mol. Microbiol. 2000, 38, 31–40.
- Fink, S.L.; Bergsbaken, T.; Cookson, B.T. Anthrax lethal toxin and Salmonella elicit the common cell death pathway of caspase-1-dependent pyroptosis via distinct mechanisms. Proc. Natl. Acad. Sci. USA 2008, 105, 4312–4317.
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Vande, W.L.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121.
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665.
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116.
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015, 25, 1285–1298.
- Yu, J.; Nagasu, H.; Murakami, T.; Hoang, H.; Broderick, L.; Hoffman, H.M.; Horng, T. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 15514–15519.
- Bergsbaken, T.; Fink, S.L.; den Hartigh, A.B.; Loomis, W.P.; Cookson, B.T. Coordinated host responses during pyroptosis: Caspase-1-dependent lysosome exocytosis and inflammatory cytokine maturation. J. Immunol. 2011, 187, 2748–2754.
- Rogers, C.; Erkes, D.A.; Nardone, A.; Aplin, A.E.; Fernandes-Alnemri, T.; Alnemri, E.S. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat. Commun. 2019, 10, 1689.
- Liu, Z.; Wang, C.; Yang, J.; Zhou, B.; Yang, R.; Ramachandran, R.; Abbott, D.W.; Xiao, T.S. Crystal Structures of the Full-Length Murine and Human Gasdermin D Reveal Mechanisms of Autoinhibition, Lipid Binding, and Oligomerization. Immunity 2019, 51, 43–49.
- Kuang, S.; Zheng, J.; Yang, H.; Li, S.; Duan, S.; Shen, Y.; Ji, C.; Gan, J.; Xu, X.W.; Li, J. Structure insight of GSDMD reveals the basis of GSDMD autoinhibition in cell pyroptosis. Proc. Natl. Acad. Sci USA 2017, 114, 10642–10647.
- Rogers, C.; Fernandes-Alnemri, T.; Mayes, L.; Alnemri, D.; Cingolani, G.; Alnemri, E.S. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun. 2017, 8, 14128.
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671.
- Aglietti, R.A.; Dueber, E.C. Recent Insights into the Molecular Mechanisms Underlying Pyroptosis and Gasdermin Family Functions. Trends Immunol. 2017, 38, 261–271.
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158.
More
Information
Subjects:
Medicine, Research & Experimental
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.5K
Revisions:
3 times
(View History)
Update Date:
21 Nov 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No