 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Peter Cragg | + 3611 word(s) | 3611 | 2020-11-25 06:57:20 | | | |
| 2 | Peter Tang | Meta information modification | 3611 | 2020-12-12 08:47:08 | | | | |
| 3 | Peter Tang | Meta information modification | 3611 | 2020-12-12 08:53:14 | | |
Video Upload Options
Calixarenes have been shown to have antimicrobial effects since the 1950s. These properties are exemplified through their applications as prodrugs, drug delivery agents and biofilm inhibitors. A particularly important development in recent years has been their ability to engage in multivalent interactions with proteins, thus inhibiting cellular aggregation.
1. Introduction
Calixarenes have found an enormous range of applications since their first discovery and later exploitation by researchers worldwide [1]. Their ability to complex small molecules, either within each individual macrocycle or through aggregated nanostructures, has opened up avenues for drug delivery. Functionalization, though upper and lower rims, offers up the possibility of their use as inhibitors of biochemical processes or as prodrugs with the active substituents released in response to external stimuli. Some useful summaries of the broader biological and biochemical effects of water-soluble calixarenes have been published for those seeking a broader perspective [2][3].
The use of calixarenes and related macrocycles as antimicrobial agents dates back to the 1950s and Cornforth's work on Macrocyclon (Figure 1) [4]. Since his original antitubercular study, the biological effects of calixarenes have become more widely appreciated. The antimicrobial impact of calixarenes was first assessed in 2002 by Regnouf-de-Vains [5] and has been followed by subsequent reviews [6][7][8]. Below, we discuss the field of antimicrobial calixarenes together with the most recent advances.
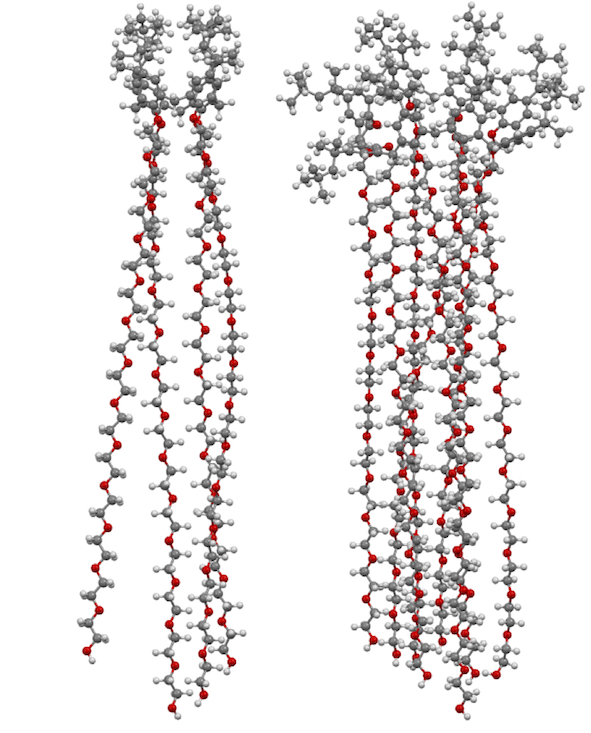
Figure 1. Macrocyclon as originally envisaged by Cornforth (left) and as later determined (right).
There are three main modes of action by which macrocycles can induce a biological response. Individual calixarenes may do so through, for example, insertion in a cell membrane to destroy its integrity, or by acting as a prodrug, incorporating a substituent that is cleaved from its parent macrocycle to become biologically active. Macrocycles, or aggregates of macrocycles, can deliver single drug molecules by transporting them in their central cavities, releasing them in response to an external stimulus. Finally, there is the possibility of disrupting the biofilms formed by colonies of bacteria when adhering to surfaces.
2. Molecular Prodrugs and Drug Delivery Agents
Calixarenes provide a rigid scaffold to which substituents can be appended through lower rim substitution reactions. The regiochemistry of these reactions can be controlled by numerous well-known techniques to maximize the distance between substituents, in alternate conformations, or make use of the macrocyclic cavity to preorganize the spatial relationships between substituents in a cone conformer. If the substituents are pharmacologically active in their own right, the macrocycle becomes the agent that delivers the prodrug to a destination within the target microbe. Once at the destination, local conditions, typically pH or enzyme action, initiate bond cleavage to release the active drug while the calixarene is either chemically degraded or excreted. Alternatively, the calixarene essentially acts as a vector by delivering its drug-derived substituents to their destination while remaining attached throughout. The benefits of such an approach are that the solubility of the macrocycle can enhance the bioavailability of the drug and that multiple copies of that drug can be delivered to the same cellular site. This latter effect is an example of multivalency and is important in other antimicrobial applications of calixarenes, notably in biofilm inhibition.
It is often only through conjugation to a carrier molecule that a drug can be delivered to its target either due to solubility issues, which are addressed by tuning the carrier's physical properties to those of its biological target, or through the affinity of the carrier for a particular feature of the target such as a phospholipid membrane or protein recognition site. This "magic bullet" approach has long been the goal of medicinal chemists, particularly when the therapeutic agents involved are highly toxic [9].
2.1. Incorporation of Antibiotic Motifs
The group of Regnouf-de-Vains has investigated the effects of introducing classical antibiotic motifs into calixarenes through research lasting over two decades. Calix[4]arenes have been used as a platform for drug delivery through the incorporation of penicillin core moieties as lower rim substituents (Figure 2). The lower rim of 4-t-butylcalix[4]arene was functionalized in the 1,3-positions to form diamide 1 [10]. Although antimicrobial data were not published, this route led the way to an analogue, 2, in which penicillin V was appended to the calixarene [11] and tested against Gram-positive and Gram-negative bacteria [12][13]. The group also prepared a nalidixic acid delivering prodrug, 3 and a fourth derivative incorporating nalidixic acid and penicillin V substituents on opposite rings, 4 [12].
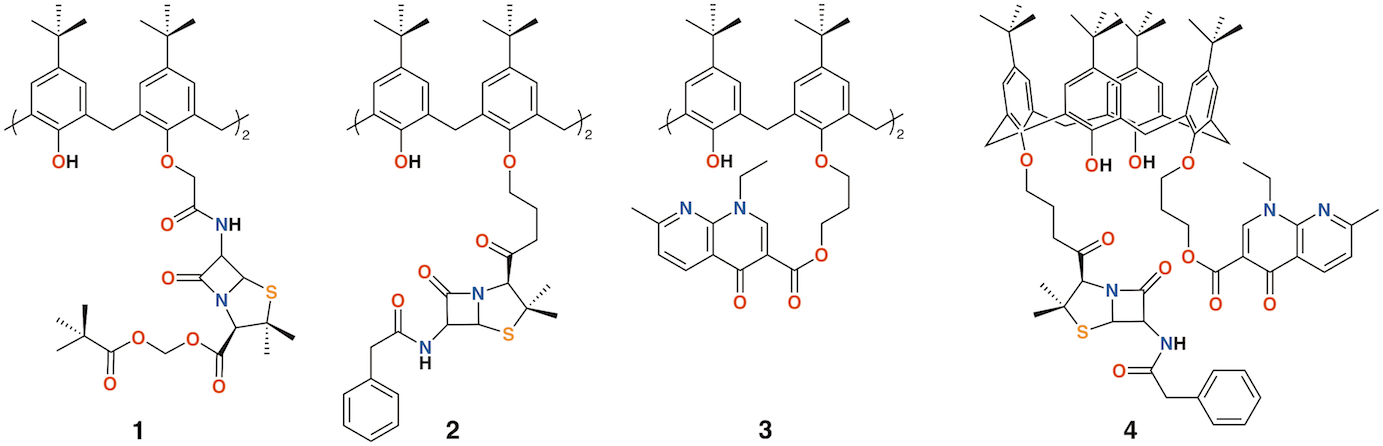
Figure 2. Regnouf-de-Vains' calixarene prodrugs.
Given the poor aqueous solubilities of these 4-t-butylcalix[4]arene derivatives, antimicrobial disc-diffusion tests were done in dimethyl sulfoxide with the pure solvent as a control and it was determined that the antimicrobial effects in all cases were due to the solvent alone. Only calixarenes 2 and 4, with two penicillin V or one penicillin V and one nalidixic acid subunit, had any significant inhibitory effect, and then only against gram-positive Staphylococcus aureus [12]. The group also explored water soluble 4-guanidinoethylcalix[4]arene derivatives 5 and 6 (Figure 3) [14][15]. Disc diffusion and minimum inhibitory concentration tests were carried out on Gram-negative Escherichia coli and Pseudomonas aeruginosa, and Gram-positive S. aureus and Enterococcus faecalis bacteria. Calix[4]arene 5 was active at 16 μg mL−1 against E. coli, S. aureus, and E. faecalis, and at 64 μg mL−1 against P. aeruginosa [16]. In 2010, the group published a more comprehensive assessment of the antimicrobial activity of 5 against 69 clinical isolates compared with two antiseptic cationic compounds, chlorhexidine and hexamidine [17]. Calixarene 6 was additionally used to treat Mycobacterium tuberculosis where it was found to have conformation-dependent minimum inhibitory concentrations (MICs) 8 μg mL−1 for the coneconformer and 1.2 μg mL−1 for the 1,3-alt conformer [18].
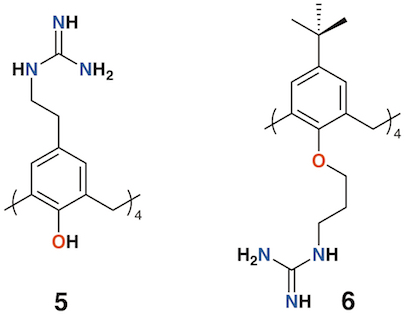
Figure 3. Regnouf-de-Vains' soluble guanidinium-functionalized calixarenes.
Pur and Dillmaghani introduced 6-aminopenicillanic acid substituents to calix[4]arene via esterification at either the lower rim, 7, or upper rim, 8, to produce "calixpenams", followed by oxidation to their sulfoxide derivatives and finally, through ring expansion and sulfur insertion, to cephalosporin-containing analogues, or "calixcephems", 9 and 10 (Figure 4). The calixpenams' MICs were found to be five to six times lower than penicillin V or X [19]. Calixpenams were effective against methicillin-sensitive S. aureus (MSSA) β-lactamase (−) whereas the calixcephems showed broad antibiotic activity. In all cases, the calixarenes were between 6 and 10 times as effective as their penicillin or cephalosporin analogues [20]. The synergistic effect of the four substituents was suggested as one reason for the enhanced activity.
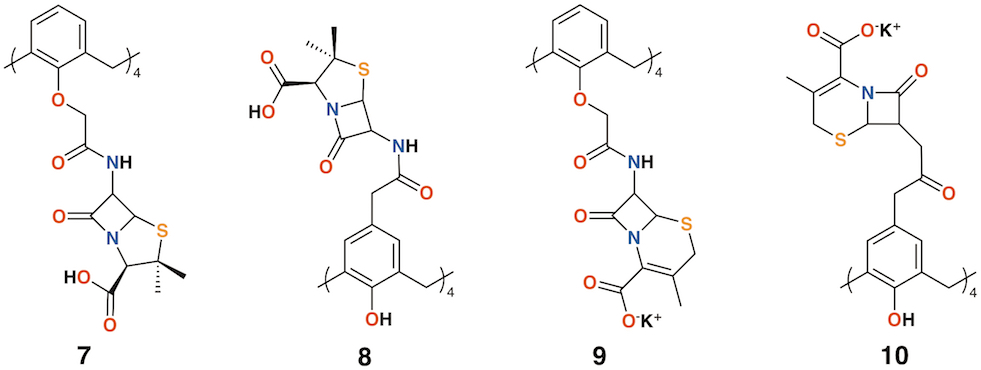
Figure 4. Dillmaghani's calixpenams and calixcephems.
2.2. Drug-Delivering Calixarenes
A polycationic calix[4]arene derivative, 11 (Figure 5), was prepared by Nostro and colleagues to deliver ofloxacin, chloramphenicol, and tetracycline [21]. The calixarene alone had a similar profile to chloramphenicol against S. aureus, but its MIC was lower by a factor of two against methicillin-resistant S. aureus (MRSA) and P. aeruginosa, and lower by a factor of four against Staphylococcus epidermidis. The authors speculate that the polycationic nature of the macrocycle may have a disruptive effect on the membrane structure of Gram-negative bacteria in a similar manner to the guanidinium derivatives discussed above.
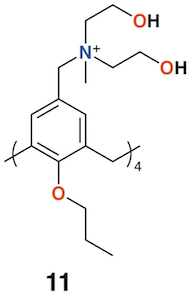
Figure 5. Nostro's drug delivering calixarene.
2.3. Metal-Binding Calixarenes
The use of calixarenes as cation-releasing platforms has been investigated by several groups, however, it is often found that the complex is no more efficacious than the parent macrocycle. There are some exceptions: Memon and colleagues used a diamide calixarene derivative, 12 (Figure 6), to complex iron(III) [22] and copper(II) [23]. Iron(III) bound in a 1:1 ratio and copper(II) in a 2:1 ratio based on spectroscopic Job's plots. Tests against bacteria and fungi found that the calixarene had MIC values between 1.5 and 3 μg mL−1, whereas the iron(III) complex had a value of 0.37 μg mL−1 against Staphylococcus albus, E. coli, and the fungal strain Rhizopus stolonifera. The copper(II) complex had the same MIC for E. coli, but was slightly less active against S. albus and R. stolonifera, with an MIC of 0.75 μg mL−1.
Shaabani and colleagues prepared a thiosemicarbazide functionalized calixarene, 13 (Figure 6), and investigated its effects, and those of its transition metal complexes, on S. aureus, Bacillus subtilis, E. coli, P. aeruginosa, Candida albicans, and Candida glabrata [24]. The metal complexes generally showed enhanced activity against E. coli, with the greatest effects seen for the nickel(II) and zinc(II) complexes, with MICs of 62.5 μg mL−1 and 31.25 μg mL−1, respectively.
The Shaabani group extended the thiosemicarbazide motif through reaction with salicylaldehyde to give 2-hydroxybenzeledene-thiosemicarbazone 14 (Figure 6) [25]. Complexes Co×14, Cu×14, Ni×14, and Zn×14 were assessed for their antibacterial effects on S. aureus, B. subtilis, E. coli, and P. aeruginosa, together with their antifungal activity on C. albicans and C. glabrata. All had MICs of 31.25 μg mL−1 against B. subtilis, E. coli, and P. aeruginosa, but only Cu×14 had any activity against S. aureus, with an MIC of 31.25 μg mL−1. Only Co×14 had any antifungal effects, and then only against C. albicans, with MICs of 31.25 μg mL−1.
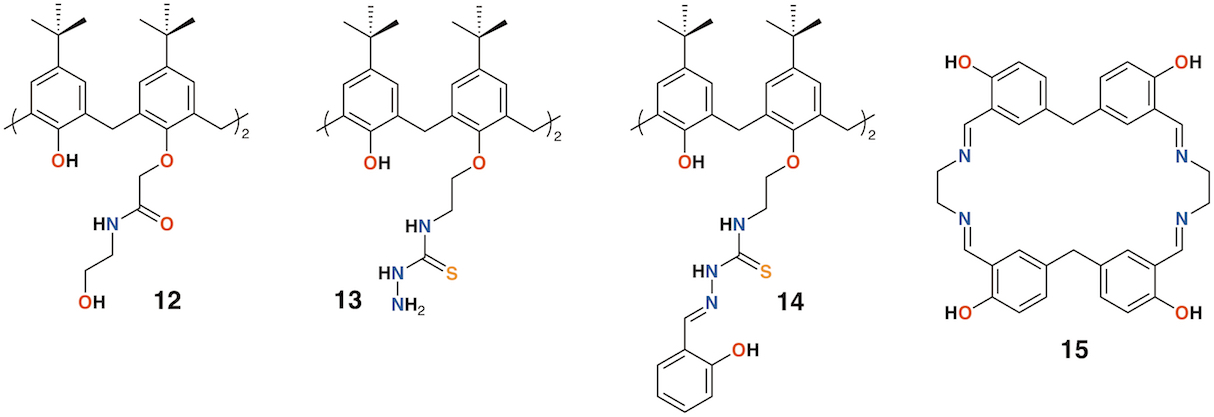
Figure 6. Cation-releasing calixarenes from Memon and Shaabani with Desai's corand.
Ray, Deolalkar, and Desai reported the synthesis of several calixarene-salen hybrid corand macrocycles, including 15, together with their silver complexes (Figure 6) [26]. The antibacterial properties of the silver complexes were assessed against E. coli, P. aeruginosa, Salmonella typhi, S. aureus, Streptococcus pyogenes, and B. subtilis. Their antifungal effects on C. albicans were also investigated. Ag×15 was significantly more potent than ampicillin against all strains tested. Of particular note was its activity against E. coli (MIC of 49.5 μg mL−1 vs. 100 μg mL−1 for ampicillin and 24.8 μg mL−1 for ciproflaxin), S. aureus (MIC of 49.5 μg mL−1 vs. 250 μg mL−1 for ampicillin or 50.0 μg mL−1 for ciproflaxin), and S. pyogenes (MIC of 79.3 μg mL−1 vs. 100 μg mL−1 for ampicillin and ciproflaxin).
3. Cell Destruction
As an alternative mode of action to drug delivery or release of toxic metal ions, the amphiphilic properties of macrocycles can be utilized to disrupt bacterial and fungal membranes, leading to rupture and cell death. Where the compounds are charged, there may be an even greater affinity for biological membranes with, potentially, different responses to Gram-positive and Gram-negative bacteria, and to fungi, which have a different membrane composition.
3.1. Macrocyclon
In 1951, Cornforth and colleagues reported on the suppressive effects of the non-ionic surfactant Triton A20 on tuberculosis in mice [27]. The surfactant consisted of a 4-(2,4,4-trimethylpentan-2-yl)phenol head group, or "octylphenol", linked to polyether substituents of varying lengths formed through reaction with ethylene oxide. The group later prepared a macrocyclic analogue of Triton A20 with methylene linkers, Macrocyclon, 16 [28]. It was essentially non-toxic to mammals and, unlike its linear analogue, found to be more potent than streptomycin in the treatment of the human virulent strain of Mycobacterium tuberculosis, H37Rv. None of the derivatives inhibited growth of tubercular bacilli and it was suggested that activity was due to their surfactant effects. In a later paper, D'Arcy Hart and colleagues revisited these compounds and compared Macrocyclon, with an average of 12.5 -CH2CH2O- repeat units and terminating in a hydroxy group, with "HOC-60", 17, an analogue with 60 repeat units (Figure 7) [29]. At this point, it was also appreciated that the compounds were calix[8]- rather than calix[4]arene derivatives. It was observed that growth of M. tuberculosis inside macrophages was inhibited by Macrocyclon but stimulated by HOC-60. Similarly, lipase activity was inhibited by Macrocyclonand stimulated by HOC-60, suggesting that lipids and lipid metabolism were affected by the macrocycles.
Almost 50 years after the original report, Macrocyclon was reinvestigated by Tascon and colleagues, who confirmed the original findings [30]. In these later experiments, both macrophages and live mice were infected with M. tuberculosis and the range of calixarenes was increased.
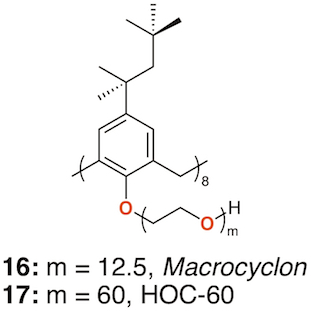
Figure 7. Cornforth's Macrocyclon and HOC-60.
Treatment with Macrocyclon supported the novel therapeutic pathway proposed by D'Arcy Hart, as the macrocycle enhanced the innate defense mechanisms in the murine macrophages. This work was followed up by a much more extensive screen of 25 calixarenes against M. tuberculosis by Hailes and colleagues [31]. Derivatives based on calix[4], -[6], -[7], and -[8]arenes with t-butyl, phenyl, and sulfonate upper rim substituents, and a range of lower rim, largely ethylene glycol substituents, were assessed alongside Macrocyclon. The parent 4-t-butylcalix[8]arene and 4-phenylcalix[8]arene were more active than their smaller homologues and the 4-sulfonatocalix[8]arene, being water soluble, had activity approaching that of Macrocyclon. The addition of polyethylene glycol substituents enhanced the calixarenes' anti-mycobacterial properties, with complete substitution having greater effects than partial substitution. Longer substituents were required for 4-phenylcalix[7]arene derivatives to be effective, presumably owing to the parent compound's lower solubility compared with the 4-t-butylcalixarenes. Lower rim acetate groups elicited pro-tubercular activity and other substituents, such as cyanopropoxy groups, had little effect.
3.2. Charged Calixarenes
Regnouf-de-Vains and colleagues investigated the anti-mycobacterial activities of 17 charged calix[4]arenes [32][33][34]. None of the anionic species exhibited activity, but the parent 4-guanidinoethylcalix[4]arene, 5 (Figure 3) , and a derivative with two lower rim bipyridyl substituents provided almost 100% inhibition against M. tuberculosis at concentrations of 1.22 μg mL−1 and 1.89 μg mL−1, respectively.
Independently, the groups of Yushchenko [35] and Loftsson [36] focused on cationic calix[4]arenes with trimethylammonium, N-(2-hydroxyethyl)-N,N-dimethylammonium, or N-(2-aminoethyl-N,N-dimethylammonium groups linked to the upper rims by methylene groups, and O-methyl, O-propyl, or O-octyl lower rim substituents, 18 to 21 (Figure 8). Derivatives 18 and 21 showed increasing hemolysis from 0.71 μg mL−1 and 0.89 μg mL−1, respectively. The antimicrobial activity of 2-hydroxyethyl derivatives 18 and 21, together with N-methylimidazolium derivatives 22and 23 (Figure 8) against S. aureus and E. coli, was studied. Compounds 18 and 22 had pronounced activity with MICs of 1.95 μg mL−1 for S. aureus while 20 and 21 were inactive. The authors concluded that the antibacterial activity depended on the size and conformational rigidity of the macrocycle and the length of the alkyl substituents on the lower rim of the calixarene. Conformationally rigid macrocycles 19 and 22 were active, while conformationally flexible 21 was inactive. In addition, with an increase in the length of the lower rim alkyl substituents from propyl to octyl, the antibacterial activity decreases.
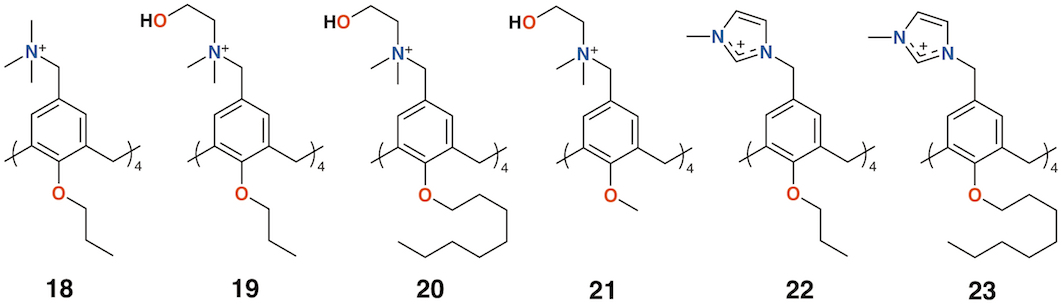
Figure 8. Charged calixarenes reported by Loftsson and Yushchenko.
3.3. Vancomycin Mimicking Calixarenes
A different approach to imparting antibiotic activity to calixarenes was pioneered by Ungaro and colleagues in 1996 [37]. Vancomycin mimics were prepared by bridging two opposite rings in O-propyl calix[4]arene through their upper rims with two, 23, or four alanine residues, 24, linked by diethylenetriamine, as shown in Figure 9. The compounds' effects on three strains of S. aureus together with Bacillus cereus, Saccharomyces cerevisiae, Acholeplasma laidlawii, S. epidermidis, E. coli and C. albicans were determined and compared to that of vancomycin. Significant activity against S. epidermidis and all strains of S. aureus, with MICs between 4 μg mL−1 and 16 μg mL−1, were found for the -D-Ala-NHCH2CH2NHCH2CH2-D-Ala- and its L-Ala analogue. These compared favorably with 2 μg mL−1 seen for vancomycin. Neither vancomycin nor the alanine-bridged calixarenes showed activity against S. cerevisiae, A. laidlawii, or C. albicans.
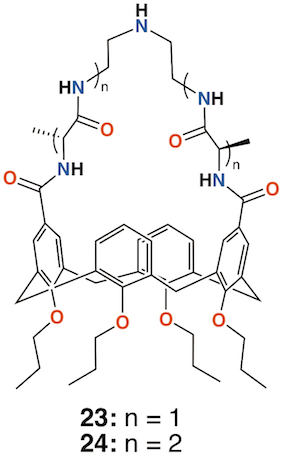
Figure 9. Ungaro's vancomycin analogues.
4. Biofilm Inhibition
Direct delivery of a drug or prodrug to individual cells is one antimicrobial strategy, however, disruption of bacterial colonies growing on a surface is another approach. Once individual bacteria are able to bind to a surface, initially through hydrophobic and other weak interactions, they can act as anchors for further cell deposition. Biofilms then form when the bacteria are able to deposit an extracellular matrix of polymeric material, largely composed of polysaccharides, which allows them to extend the colony [38]. The polysaccharides are recognized by lectins, carbohydrate-binding proteins present on cell surfaces, leading to the formation of a cellular matrix. These complex, surface-bound films accumulate more bacteria, facilitating chemical communication between them. One consequence of this is increased resistance to antibiotics and surfactant-based detergents as the outer layers form a protective barrier for the remainder of the colony. While the biofilm may comprise a single species, it is possible for synergistic ecosystems to emerge in which molecules generated by one species are metabolized by others. As different species of bacteria have different susceptibilities to antibiotics, biofilm heterogeneity can also confer a level of overall protection.
While antibiotics and surfactants are widely used against biofilms, inhibition of their formation and growth has been approached using multivalent macrocycles. Several groups have developed derivatives with extended substituents terminating in saccharides such as mannose [39][40]. The sugar termini bind to receptor sites on bacterial proteins. These sites would usually interact with polysaccharide groups in the extracellular matrix, so blocking them inhibits adhesion. Calixarenes incorporating several substituents have been developed as inhibitors. A single macrocycle with several sugar groups has two benefits. Its multivalency maximizes the chances that it will block binding sites and extended substituents are able to span between several receptor sites on the same protein. In doing so, they can form a physical barrier between the protein surface and the extracellular matrix. Consequently, the conformation of the calixarene is often a vital factor in the compound's effectiveness. The cone conformer of calix[4]arene is able to bind four sites on the same protein face and is essentially tetravalent, whereas the same compound in the 1,3-alt conformer would have a greater span between binding sites, but would only be divalent.
Lectin binding by macrocyclic glycoconjugates can also reduce infection by stopping the cell adhesion process from the very beginning. With carbohydrate recognition sites blocked, bacteria have to rely on weak protein–surface interactions to aggregate and these are easily reversed. To determine the efficacy of the macrocyclic agents, two key factors need to be quantified: affinity and inhibition. Experiments undertaken in the presence of different lectins will reveal specificity and affinity, while biofilm inhibition properties are evaluated through cell culture experiments.
4.1. Calixsugars
In 2009, Imberty, Matthews, Vidal, and colleagues prepared a family of 4-t-butylcalix[4]arenes with galactose substituents attached by a triethylene glycol tether using click chemistry [41]. The fully substituted derivatives gave rise to cone, 1,3-alt, and partial cone products 25 to 27(Figure 10). The tetrasubstituted mannose analogue, 28 (Figure 10), was also prepared, in the cone conformer, as a comparator. Binding to Lec A (galactophilic P. aeruginosa first lectin, or PA-IL) occurred with the tetravalent galactose ligands binding most strongly particularly when in the partial cone and 1,3-alt conformers. This demonstrated the importance of geometric alignment when binding to the protein surface. Imberty, Vidal, and colleagues then investigated the influence of the linker arm between the calixarene and sugar terminus [42]. Linkers with increasing rigidity but similar lengths were compared with the original triethylene glycol linker. The cost of increasing rigidity was lower solubility, nevertheless, the researchers found that the calixarene with four diethylene glycol acetamide linkers was more potent than the original triethylene glycol-containing compounds, but only when in the 1,3-alt conformation.
Using a similar, but simpler approach, Consoli, Geraci, and colleagues synthesized a tetra-O-propyl cone-calix[4]arene, 29 (Figure 10) with fucose groups linker to the upper rim [43]. Biofilm inhibition was observed for P. aeruginosa with a dose-dependent response seen over the range from 2.0 μg mL−1 to 32.7 μg mL−1 and concomitant inhibition rates rising from 35% to 73%. The authors ascribe this to interactions between the bacteria and the formation of positively charged ammonium groups.
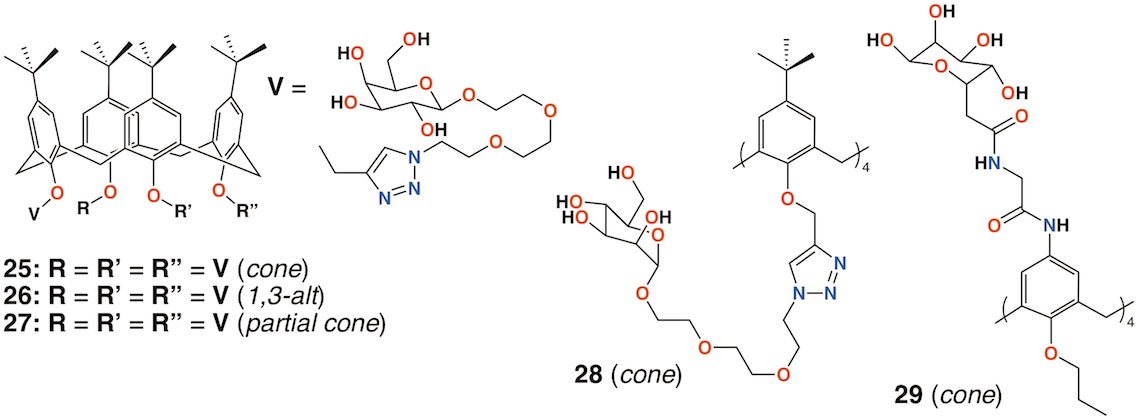
Figure 10. Glycosylated calixarenes of Imberty and Geraci.
Vidal and colleagues extended the family of calixarenes to include those with four fucose termini, 30 (Figure 11) [44]. With glucose, galactose, mannose, and fucose analogues in hand, their effects on bacterial aggregation, cell adhesion, and biofilm inhibition could be compared. As LecA is selective for galactose and LecB (fucophilic P. aeruginosa second lectin, or PA-IIL), discrimination between the two should prove possible. The data suggested that a 1,3-alt calixarene with four fucose-terminated substituents appeared to utilize three of them when binding and its affinity for LecB was 50 times lower. The glucosylated analogue was not recognized by either lectin.
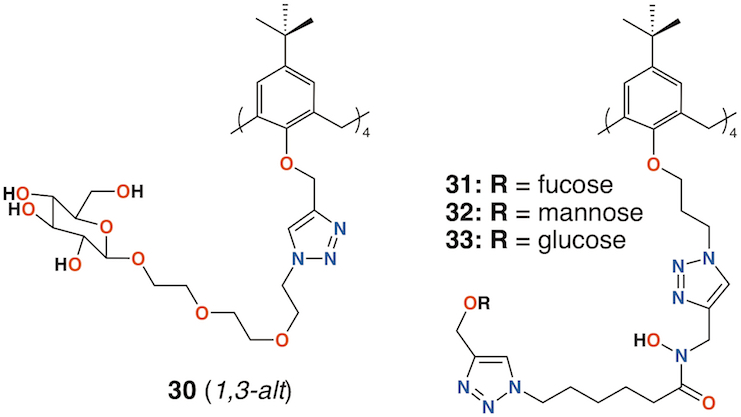
Figure 11. Glycosylated calixarenes of Vidal and Benazza.
Benazza and colleagues prepared fuco-, 31; manno-, 32; and glucocluster, 33, cone-calix[4]arene derivatives, shown in Figure 11, and introduced an iron chelating hydroxamic acid region in each linker [45]. It was proposed that, in addition to binding to lectins, these compounds could sequester soluble iron, an essential nutrient for bacteria, to enhance their antibacterial activities. No antibiotic activity against P. aeruginosa was seen for the fucose or mannose terminated derivatives, so additional experiments investigated the effects of iron chelation. Under iron-depleted conditions, production of the bacteria's main siderophore, pyoverdine-I (Pvd-I), increased to counteract the sequestering effects of the calixarenes. Binding assays revealed an unexpected interaction between LecB and the mannosylated calixarene to be over 200 times stronger than a monomeric analogue.
Computer modelling indicated that the calixarene could bind simultaneously to four sites on the LecB protein, as shown in Figure 12, which may explain this effect. P. aeruginosa biofilm growth studies at 5.4 μg mL−1 found that not only did the mannosylated-derivative inhibit biofilm formation by 84%, but the fucoslyated-derivative did so by 72%. Even more surprising was the effect of the glucosylated-derivative, used as a negative control, which gave 92% inhibition. The authors concluded that other inhibitory mechanisms could be involved such as the possibility that nitric oxide was being released from the iron-binding site.
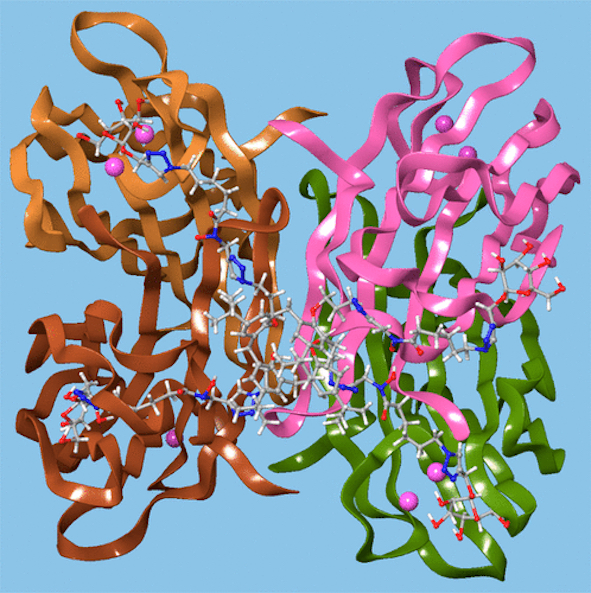
Figure 12. A glycosylated calixarene binding to a lectin. Reprinted with permission from [45]. Copyright © 2019 American Chemical Society.
5. Conclusions
From Cornforth's O-alkylation of calixarenes with cell membrane-disrupting polyethers, to Ungaro's vancomycin mimics through to Regnouf-de-Vains' nalidixic acid-appended calixarene prodrugs, and Matthews' multivalent lectin-binding calixarenes, calixarenes and their related macrocycles have been demonstrating antimicrobial activity for almost 70 years. During this time, the modes of action have evolved from simple surfactant activity through to drug delivery and, most recently, the use of multivalent macrocycles to bind proteins to block their aggregation. Indeed, it is the chemist's ability to prepare complex multifunctional derivatives with precise regioisomerism, which is propelling the most recent advances in antimicrobial macrocycles. Control of molecular recognition over extended distances is key in this endeavor and yet it relies on some of the first aspects of calixarene chemistry to be exploited chemically: lower rim substitution and conformer control.
Several themes are beginning to emerge: calixarenes incorporating drug moieties such as penicillin are more effective when coupled to ammonium or guanidinium substituents; metal chelating and releasing calixarenes are not as effective as might be expected; and the ability to use macrocycles as multivalent ligands capable of binding in spatially remote locations has been shown to be a highly effective biofilm-disrupting strategy. Future development will, no doubt, revolve around substituents with greater specificity for target binding sites, such that the compounds are effective at vanishingly small doses.
References
- Neri, P.; Sessler, J.L.; Wang, M.-X. (Eds.) Calixarenes and Beyond; Springer International Publishing: Cham, Switzerland, 2016.
- Perret F.; Lazar, A.N.; Coleman, A.W. Biochemistry of the para-sulfonato-calix[n]arenes. Chem. Commun. 2006, 2425–2438.
- Perret F.; Coleman, A.W. Biochemistry of anionic calix[n]arenes. Chem. Commun. 2011, 47, 7303–7319.
- Cornforth, J.W.; D’Arcy Hart, P.; Nicholls, G.A.; Rees, R.J.W.; Stock, J.A. Antituberculous effects of certain surface-active polyoxyethylene ethers. J. Pharmacol. 1955, 10, 73–86.
- Lamartine, R.; Tsukada, M.; Wilson, D.; Shirata, A. Antimicrobial activity of calixarenes. Comptes Rendus Chim. 2002, 5, 163–169.
- Naseer, M.M.; Ahmed, M.; Hameed, S. Functionalized calix[4]arenes as potential therapeutic agents. Biol. Drug Des. 2017, 89, 243–256.
- Rodik, R.; Poberezhnyk, M.; Kalchenko, V. Calixarene derivatives for (nano)biotechnologies. Macroheterocycles 2017, 10, 421–431.
- Rodik, R.V. Антимікробна та Противірусна Активність Каліксаренів. Org. Pharmaceut. Chem. 2015, 13, 67–78.
- Strebhardt, K.; Ullrich, A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Rev. Cancer 2008, 8, 473–480.
- Ben Salem, A.; Regnouf-de-Vains, J.-B. Synthesis and characterisation of a new podand based on a calixarene and a β-lactam. Tetrahedron Lett. 2001, 42, 7033–7036.
- Korchowiec, B.; Ben Salem, A.; Corvis, Y.; Regnouf-de-Vains, J.-B.; Korchowiec, J.: Rogalska, E. Calixarenes in a membrane environment: A monolayer study on the miscibility of three p-tert-butylcalix[4]arene b-lactam derivatives with 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine. Phys. Chem. B2007, 111, 13231–13242.
- Ben Salem, A.; Sautrey, G.; Fontanay, S.; Duval, R.E.; Regnouf-de-Vains, J.-B. Molecular drug-organiser: Synthesis, characterization and biological evaluation of penicillin V and/or nalidixic acid calixarene-based podands. Med. Chem. 2011, 19, 7534–7540.
- Ben Salem, A.; Regnouf-de-Vains, J.-B. Towards a new family of calixarene-based podands incorporating quinolone arms. An example using nalidixic acid.Tetrahedron Lett. 2003, 44, 6769–5771.
- Gutsche, C.D.; Nam, K.C. Calixarenes. 22. Synthesis, properties, and metal complexation of aminocalixarenes. J. Am. Chem. Soc. 1988, 110, 6153–6162.
- Baker, T.J.; Tomioka, M.; Goodman, M.; Mergott, D.G.; Roush, W.R. Preparation and use of N,N′-di-BOC-N″-triflylguanidine. Synth. 2000, 78, 91.
- Mourer, M.; Duval, R.E.; Finance, C.; Regnouf-de-Vains, J.-B. Functional organisation and gain of activity: The case of the antibacterial tetra-para-guanidinoethyl-calix[4]arene. Med. Chem. Lett. 2006, 16, 2960–2963.
- Grare, M.; Massimba Dibama, H.; Lafosse, S.; Ribon, A.; Mourer, M.; Regnouf-de-Vains, J.-B.; Duval, R.E. Cationic compounds with activity against multidrug-resistant bacteria: Interest of a new compound compared with two older antiseptics, hexamidine and chlorhexidine. Microbiol. Infect. 2010, 16, 432–438.
- Mourer, M.; Duval, R.E.; Constant, P.; Daffé, M.; Regnouf-de-Vains, J.-B. Impact of tetracationic calix[4]arene conformation-from conic structure to expanded bolaform-on their antibacterial and antimycobacterial activities. ChemBioChem 2019, 20, 911–921.
- Pur, F.N.; Dilmaghani, K.A. Calixpenams: Synthesis, characterization, and biological evaluation of penicillins V and X clustered by calixarene scaffold. J. Chem. 2014, 38, 288–296.
- Pur, F.N.; Dilmaghani, K.A. Calixcephems: Clustered cephalosporins analogous to calixpenams as novel potential anti-MRSA agents. J. Chem. 2014, 38, 850–858.
- Consoli, G.M.L.; Granata, G.; Picciotto, R.; Blanco, A.R.; Geraci, C.; Marino, A.; Nostro, A. Design, synthesis and antibacterial evaluation of a polycationic calix[4]arene derivative alone and in combination with antibiotics. Chem. Commun. 2018, 9, 160–164.
- Memon, S.; Chandio, A.A.; Memon, A.A.; Nizamani, S.M.; Bhatti, A.A.; Brohi, N.A. Synthesis, characterization, and exploration of antimicrobial activity of copper complex of diamide derivative of p-tert-butylcalix[4]arene. Aromat. Compd. 2017, 37, 362–374.
- Chandio, A.A.; Memon, A.A.; Memon, S.; Memon, F.N.; Panhwar, Q.K.; Durmaz, F.; Nizamani, S.M.; Brohi, N.A. Synthesis and antimicrobial assessment of Fe3+ inclusion complex of p-tert-butylcalix[4]arene diamide derivative. Chem. 2019, 2019, doi:10.1155/2019/2534072.
- Noruzi, E.B.; Kheirkhahi, M.; Shaabani, B.; Geremia, S.; Hickey, N.; Asaro, F.; Nitti, P.; Kafil, H.S. Design of a thiosemicarbazide functionalized calix [4] arene ligand and related transition metal complexes: Synthesis, characterization and biological studies. Chem. 2019, 7, 663.
- Noruzi, E.B.; Shaabani, B.; Geremia, S.; Hickey, N.; Nitti, P.; Kafil, H.S. Synthesis, crystal structure, and biological activity of a multidentate calix[4]arene ligand doubly functionalized by 2-hydroxybenzeledene-thiosemicarbazone. Molecules 2020, 25, 370.
- Roy, H.; Deolalkar, M.; Desai, A.S. Synthesis of calix-salen silver corates for evaluation of their antimicrobial and anticancer activities. ACS Omega 2019, 4, 21346–21352.
- Cornforth, J.W.; D’Arcy Hart, P.; Rees, R.J.W.; Stock, J.A. Antituberculous effect of certain surface-active polyoxyethylene ethers in mice. Nature 1951, 168, 150–153.
- Zinke, A.; Zigeuner, G.; Hössinger, K.; Hoffmann, G. Zur Kenntnis des Härtungsprozesses von Phenol-Formaldehyd-Harzen. XVIII., vorläufige Mitteilung: Über cyclische Mehrkernphenole. Chem. 1948, 79, 438–439.
- D’Arcy Hart, P.; Armstrong, J.A.; Brodaty, E. Calixarenes with host-mediated potency in experimental tuberculosis: Further evidence that macrophage lipids are involved in their mechanism of action. Immun. 1996, 64, 1491–1493.
- Colston, M.J.; Hailes, H.C.; Stavropoulos, E.; Hervé, A.C.; Hervé, G.: Goodworth, K.J.; Hill, A.M.; Jenner, P.; D’Arcy Hart, P.; Tascon, R.E. Antimycobacterial calixarenes enhance innate defense mechanisms in murine macrophages and induce control of Mycobacterium tuberculosis infection in mice. Immun. 2004, 72, 6318–6323.
- Goodworth, K.J.; Hervé, A.C.; Stavropoulos, E.; Hervé, G.; Casades, I.; Hill, A.M.; Weingarten, G.G.; Tascon, R.E.; Colston, M.J.; Hailes, H.C. Synthesis and in vivo biological activity of large-ringed calixarenes against Mycobacterium tuberculosis. Tetrahedron 2011, 67, 373–382.
- Mourer, M.; Massimba Dibama, H.; Constant, P.; Daffé, M.; Regnouf-de-Vains, J.-B. Anti-mycobacterial activities of some cationic and anionic calix[4]arene derivatives. Med. Chem. 2012, 20, 2035–2041.
- Mourer, M.; Massimba Dibama, H.; Fontanay, S.; Grare, M.; Duval, R.E.; Finance, C.; Regnouf-de-Vains, J.-B. p-Guanidinoethyl calixarene and parent phenol derivatives exhibiting antibacterial activities. Synthesis and biological evaluation. Med. Chem. 2009, 17, 5496–5509.
- Mourer, M.; Fontanay, S.; Duval, R.E.; Regnouf-de-Vains, J.-B. Synthesis, characterization, and biological evaluation as antibacterial agents of water-soluble calix[4]arenes and phenol derivatives incorporating carboxylate groups. Chim. Acta 2012, 95, 1373–1386.
- Yushchenko, T.I.; Germanyuk, T.A.; Chornoknyzhny, S.I.; Zaichko, N.V.; Korol, A.P.; Prokopchuk, Z.M.; Rodik, R.V.; Cheshun, E.A. Antibacterial and antiplatelet activity of calix[4,6]arene tetraalkylamines. Drug Toxicol. 2012, 5, 79–88.
- Ukhatskaya, E.V.; Kurkov, S.V.; Hjálmarsdóttir, M.A.; Karginov, V.A.; Matthews, S.E.; Rodik, R.V.; Kalchenko, V.I.; Loftsson, T. Cationic quaternized aminocalix[4]arenes: Cytotoxicity, haemolytic and antibacterial activities. J. Pharm. 2013, 458, 25–30.
- Casnati, A.; Fabbi, M.; Pelizzi, N.; Pochini, A.; Sansone, F.; Ungaro, R. Synthesis, antimicrobial activity and binding properties of calix[4]arene based vancomycin mimics. Med. Chem. Lett. 1996, 6, 2699–2704.
- Flemming, H.-C.; Wingender, J. The biofilm matrix. Rev. Microbiol. 2010, 8, 623–633.
- Matthews, S.E. Calixsugars: Finally reaching their potential? In Calixarenes and Beyond; Neri, P., Sessler, J.L., Wang, M.-X., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 559–600.
- Baldini, L.; Casnati, A.; Sansone, F.; Ungaro, R. Peptido- and glycocalixarenes. In Calixarenes in the Nanoworld; Vicens, J., Harrowfield, J., Baklouti, L., Eds.; Springer: Dordrecht, The Netherlands, 2007; pp. 233–257.
- Cecioni, S.; Lalor, R.; Blanchard, B.; Praly, J.-P.; Imberty, A.; Matthews, S.E.; Vidal, S. Achieving high affinity towards a bacterial lectin through multivalent topological isomers of calix[4]arene glycoconjugates. Eur. J. 2009, 15, 13232–13240.
- Cecioni, S.; Praly, J.-P.; Matthews, S.E.; Wimmerová, M.; Imberty, A.; Vidal, S.; Rational design and synthesis of optimized glycoclusters for multivalent lectin–carbohydrate interactions: Influence of the linker arm. Eur. J. 2012, 18, 6250–6263.
- Consoli, G.M.L.; Granata, G.; Cafiso, V.; Stefani, S.; Geraci, C. Multivalent calixarene-based C-fucosyl derivative: A new Pseudomonas aeruginosa biofilm inhibitor. Tetrahedron Lett. 2011, 52, 5831–5834.
- Boukerb, A.M.; Rousset, A.; Galanos, N.; Meár, J.-B.; Theṕaut, M.; Grandjean, T.; Gillon, E.; Cecioni, S.; Abderrahmen, C.; Faure, K.; et al. Antiadhesive properties of glycoclusters against Pseudomonas aeruginosa lung infection. Med. Chem. 2014, 57, 10275–10289.
- Taouai, M.; Chakroun, K.; Sommer, R.; Michaud, G.; Giacalone, D.; Ben Maaouia, M.A.; Vallin-Butruille, A.; Mathiron, D.; Abidi, R.; Darbre, T.; et al. Glycocluster tetrahydroxamic acids exhibiting unprecedented inhibition of Pseudomonas aeruginosa J. Med. Chem. 2019, 62, 7722−7738.

