 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Federica Finetti | + 1511 word(s) | 1511 | 2020-12-09 04:34:47 | | | |
| 2 | Federica Finetti | -21 word(s) | 1490 | 2020-12-11 14:30:28 | | |
Video Upload Options
The involvement of inflammation in cancer progression has been the subject of research for many years. Inflammatory milieu and immune response are associated with cancer progression and recurrence. In different types of tumors, growth and metastatic phenotype characterized by the epithelial mesenchymal transition (EMT) process, stemness, and angiogenesis, are increasingly associated with intrinsic or extrinsic inflammation. Among the inflammatory mediators, prostaglandin E2 (PGE2) supports epithelial tumor aggressiveness by several mechanisms, including growth promotion, escape from apoptosis, transactivation of tyrosine kinase growth factor receptors, and induction of angiogenesis. Moreover, PGE2 is an important player in the tumor microenvironment, where it suppresses antitumor immunity and regulates tumor immune evasion, leading to increased tumoral progression.
1. Introduction
The involvement of inflammation in cancer progression was first described in 1863 by Rudolf Virchow. He observed that infiltrated immune cells reflect the place where cancer lesions appear in the inflamed tissue and hypothesized that chronic inflammation is a condition that predisposes one to cancer development. Most recent observations revealed that there is a direct causal link between inflammation and cancer: it is estimated that primary infections (such as Helicobacter pylori, hepatitis B and C viruses) and inflammatory responses are linked to 7% to 30% of cancer deaths worldwide [1][2]. Consistently, epidemiological observation showed that daily aspirin or other non-steroidal anti-inflammatory drugs (NSAIDs) reduced deaths due to several common cancers [3][4][5][6][7], indicating that cyclooxygenase (COX) inhibition and the reduction of its main metabolic product, prostaglandin E2 (PGE2), may prevent solid-organ cancers.
2. Prostaglandin E2 Biosynthesis and Functions
Several biological activities have been attributed to PGE2 both in physiological and pathological conditions. In physiological processes, PGE2 regulates fever, kidney function, pain, mucosal integrity, blood vessel homeostasis, and inflammation. In pathological conditions, as in cancer, PGE2 is produced by cancerous stromal cells and enhances tumor cell proliferation and survival, promotes angiogenesis, and induces metastasis. During tumor progression, PGE2 exerts its activity through ligation with four E-type prostanoid (EP) receptors 1–4 (EP 1–4), by acting on releasing cells (autocrine mechanism) and neighboring cells (paracrine mechanism) [8].
PGE2 belongs to the prostanoid family of lipids, a subclass of eicosanoids produced by oxidation of 20-carbon essential fatty acids that are localized within cell membranes. Prostanoids are synthesized by sequential actions of different and highly specific enzymes. Their synthesis is initiated after the release of arachidonic acid (AA) from membrane lipids by phospholipases A2 (PLA2) family members.
Membrane-released AA is rapidly oxidized into the unstable metabolite, prostaglandin G2 (PGG2), which is subsequently reduced to PGH2. Both steps are sequentially catalyzed by the COX enzymes. COX-1 is constitutively expressed at basal levels in many cells, generating low levels of PGs that are cytoprotective and maintain body homeostasis. In contrast, COX-2 is normally absent in most cells and it is induced in response to a variety of stimuli including growth factors and cytokines [9][10][11]. Once synthesized, PGH2 is rapidly converted into PGE2 by three distinct terminal synthases (Figure 1).
These synthases include microsomal PGE synthase-1 (mPGES-1), mPGES-2, and cytosolic PGE synthase (cPGES) and are tightly regulated under various conditions. mPGES-1 is frequently induced concomitantly with COX-2 by several proinflammatory stimuli to generate a transient increase of PGE2 levels [12][13]. The levels of PGE2 can also be regulated by its metabolic turnover. The activation of two key catabolic enzymes, 15-hydroxyprostaglandin dehydrogenase (15-PGDH) and 15-ketoprostaglandin-13-reductase (13-PGR), can essentially eliminate the biological activity of PGE2 [14].
Following synthesis, the physiological activity of PGE2 is mediated by the activation of downstream signaling cascades via seven transmembrane G-protein coupled receptors (GPCRs), referred as the EP receptors. The EP receptor family consists of four isoforms (EP1-4) coupled to heterotrimeric G proteins containing stimulatory (GαS) or inhibitory (Gαi) subunits that can modulate the levels of Ca2+, cyclic AMP (cAMP), and inositol phosphate, thus, activating divergent downstream signaling pathways [15] (Figure 1). The interaction between PGE2 and EP receptors is dependent on cell and tissue type and location. On cancer cells, the expression and localization of EP receptors may be variable and may influence cell response to PGE2. Specific effects depending on the activation of the different EP receptor subtype have been described, including EP1-dependent tumor cell migration and invasion, EP2-induced angiogenesis and suppression of the anti-tumor immune response, and, finally, EP4-related tumor cell migration and metastasis. The role of the EP3 receptor has yet to be clarified [15].
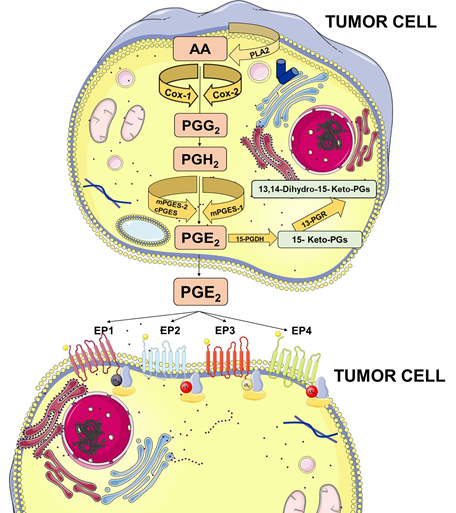
Figure 1. Prostaglandin E2 (PGE2) biosynthesis. Arachidonic acid (AA) is a polyunsaturated fatty acid that constitutes the phospholipid domain of most cell membranes and is released from the cellular membranes by cytoplasmic phospholipases A2 (PLA2). Free AA can be metabolized to PGE2 through the cyclooxygenase (COX) pathway. In this pathway, the key step is the enzymatic conversion of AA to the intermediate prostaglandin G2 (PGG2), which is then reduced to the intermediate PGH2 by the peroxidase activity of COX. PGH2 is sequentially metabolized to PGE2 by specific PGE synthases (cytosolic PGE synthase (cPGES), microsomal PGE synthase-1 (mPGES1), and mPGES2). PGE2 exerts its effects through ligation with four G-protein-coupled receptors (GPCRs), EP1–EP4. Each E-type prostanoid (EP) receptor couples to distinct signaling pathways. This figure was created using Servier Medical Art templates, which are licensed under a Creative Commons Attribution 3.0 Unported License; https://smart.servier.com.
3. Prostaglandin E2 and Cancer
PGE2 is the most abundant prostaglandin that has been found in various human malignancies. Inhibition of its production by unselective COX inhibitors such as aspirin or other NSAIDs have been associated with a reduced risk of colon, breast, lung, prostate, and other solid cancers and their recurrence (see Table 1) [16][17][18]. Furthermore, it has been reported that COX-2 and/or mPGES-1 are constitutively expressed in several cancers, including non-small cell lung cancer [19][20], colorectal cancer [21][22], breast cancer [23][24], prostate cancer [25][26], melanoma [27], and hepatocellular carcinoma [28][29], suggesting that the COX-2/mPGES-1/PGE2 pathway is linked to the neoplastic progression. To outline the importance of this pathway, several efforts have been conducted to develop selective COX-2 inhibitors lacking the side effects of unselective COX inhibitors and provided with specific activities [30]. Among these, celecoxib showed antitumoral activity being able to reduce the risk of colon, breast, prostate, and lung cancer [31]. However, long-term consumption of COX-2 inhibitors presents important side effects [30] and their use as anticancer agents has to be better investigated.
Several animal models have been developed and used to assess the efficacy of COX inhibition in vivo by using low-dose aspirin or NSAIDs, and to elucidate the molecular mechanisms of PGE2-induced tumor progression.
A large number of studies have been conducted to reveal the role of PGE2 in colon cancer carcinogenesis and progression. For example, regression of small intestinal adenomas in ApcMin/+ mice induced by NSAIDs is blocked by PGE2 treatment [32]. Moreover, the adenoma-preventive activity of celecoxib is abrogated in 15-PGDH KO mice that possess increased endogenous PGE2 levels [33]. Genetic deletion of 15-PGDH increases endogenous PGE2 levels and promotes colon tumor growth also in ApcMin/+ and azoxymethane (AOM) mouse models [34]. In addition, intraperitoneal PGE2 treatment boosts the AOM-induced colon tumor incidence and multiplicity and significantly increases proliferative index and reduces apoptotic index [35]. Likewise, it has been demonstrated in three different mouse models of intestinal tumorigenesis that chronic low-dose aspirin prevents tumor formation and that the aspirin antitumor effect is most pronounced when treatment is started before tumor initiation [36].
To further outline the important role of PGE2 in colon carcinogenesis, different studies showed that mPGES-1 deletion suppresses the development of intestinal tumors in ApcMin/+ and AOM models [37][38]. Moreover, Sasaki et al. have reported that mPGES-1 deletion reduces AOM-induced colon polyp and aberrant crypt foci (ACF) formation [39]. Similar results were reported in animal models of breast cancer [40].
Additional information regarding the role of PGE2 signaling has been obtained from xenograft animal models, in which reduction of PGE2 production results in decreased tumor growth [15][26][41][42].
It is now well accepted that a typical hallmark of tumors is an important immunosuppressive niche composed of suppressive immune cells which play a major role in the regulation of tumor progression, supporting cancer stemness and helping the tumor in the metastatic process. In the last decade, therapeutic strategies targeting these cellular populations have been developed and found to be beneficial in clinical practice (e.g., anti-PD1 and anti-CTLA4 therapies) [43]. The immune subsets that orchestrate tumor immunosuppression include myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), dendritic cells (DCs), natural killer (NK) T cells, and regulatory T-cells (Tregs). Herein, as depicted in Figure 2, the major roles of PGE2 in the fine regulation of these cells are summarized, with the aim of immunoregulation.

Figure 2. PGE2 roles in immunosuppressive tumoral niche. PGE2 levels are increased in the tumoral microenvironment. PGE2 is secreted by cancer cells and immune cells. In the figure are depicted the main functions of PGE2 in myeloid-derived suppressor cells, tumor-associated macrophages, dendritic cells, natural killer (NK) cells, and T-cells. Figure has been generated using BioRender.
This review has highlighted the pleiotropic role of PGE2 in controlling tumor cells but also the tumor microenvironment, suggesting that targeting PGE2 could be a good strategy to act on tumor progression.
References
- De Martel, C.; Ferlay, J.; Franceschi, S.; Vignat, J.; Bray, F.; Forman, D.; Plummer, M. Global burden of cancers attributable to infections in 2008: A review and synthetic analysis. Lancet Oncol. 2012, 13, 607–615.
- Schottenfeld, D.; Beebe-Dimmer, J. The cancer burden attributable to biologic agents. Ann. Epidemiol. 2015, 25, 183–187.
- Rothwell, P.M.; Fowkes, F.G.R.; Belch, J.F.; Ogawa, H.;Warlow, C.P.; Meade, T.W. E ect of daily aspirin on long-term risk of death due to cancer: Analysis of individual patient data from randomised trials. Lancet 2011, 377, 31–41.
- Cho, M.H.; Yoo, T.G.; Jeong, S.-M.; Shin, D.W. Association of aspirin, metformin, and statin use with gastric cancer incidence and mortality: A nationwide cohort study. Cancer Prev. Res. 2020.
- Lin, Y.S.; Yeh, C.C.; Huang, S.F.; Chou, Y.S.; Kuo, L.T.; Sung, F.C.; Muo, C.H.; Su, C.T.; Su, F.H. Aspirin associated with risk reduction of secondary primary cancer for patients with head and neck cancer: A population-based analysis. PLoS ONE 2018, 13, e0199014.
- Ma, Y.; Brusselaers, N. Maintenance use of aspirin or other non-steroidal anti-inflammatory drugs (NSAIDs) and prostate cancer risk. Prostate Cancer Prostatic Dis. 2018, 21, 147–152.
- Veettil, S.K.; Lim, K.G.; Ching, S.M.; Saokaew, S.; Phisalprapa, P.; Chaiyakunapruk, N. E ects of aspirin and non-aspirin nonsteroidal anti-inflammatory drugs on the incidence of recurrent colorectal adenomas: Asystematic review with meta-analysis and trial sequential analysis of randomized clinical trials. BMC Cancer 2017, 17, 763.
- Donnini, S.; Finetti, F.; Terzuoli, E.; Bazzani, L.; Ziche, M. Targeting PGE2 signaling in tumor progression and angiogenesis. For. Immunopathol. Dis. Therap. 2014.
- Dubois, R.N.; Abramson, S.B.; Cro ord, L.; Gupta, R.A.; Simon, L.S.; van de Putte, L.B.A.; Lipsky, P.E. Cyclooxygenase in Biology and Disease. FASEB J. 1998, 12, 1063–1073.
- Simmons, D.L.; Botting, R.M.; Hla, T. Cyclooxygenase Isozymes: The Biology of Prostaglandin Synthesis and inhibition. Pharmacol. Rev. 2004, 56, 387–437.
- Wang, D.; Dubois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193.
- Murakami, M.; Nakatani, Y.; Tanioka, T.; Kudo, I.; Synthase, P.E. Molecular Biology of the Arachidonate Cascade. Prostaglandins Other Lipid Mediat. 2002, 68–69, 383–399.
- Samuelsson, B.; Morgenstern, R.; Jakobsson, J. Membrane Prostaglandin E Synthase-1: A Novel Therapeutic Target. Pharmacol. Rev. 2007, 59, 207–224.
- Tai, H.H. Prostaglandin catabolic enzymes as tumor suppressors. Cancer Metastasis Rev. 2011, 30, 409–417.
- O’Callaghan, G.; Houston, A. Prostaglandin E2 and the EP receptors in malignancy: Possible therapeutic targets? Br. J. Pharmacol. 2015, 172, 5239–5250.
- Cai, Y.; Yousef, A.; Grandis, J.R.; Johnson, D.E. NSAID therapy for PIK3CA-Altered colorectal, breast, and head and neck cancer. Adv. Biol. Regul. 2020, 75, 100653.
- Harris, R.E.; Beebe-Donk, J.; Doss, H.; Doss, D.B. Aspirin, ibuprofen, and other non-steroidal anti-inflammatory drugs in cancer prevention: A critical review of non-selective COX-2 blockade (review). Oncol. Rep. 2005, 13, 559–583.
- Drew, D.A.; Schuck, M.M.; Magicheva-Gupta, M.V.; Stewart, K.O.; Gilpin, K.K.; Miller, P.; Parziale, M.P.; Pond, E.N.; Takacsi-Nagy, O.; Zerjav, D.C.; et al. E ect of Low-dose and Standard-dose Aspirin on PGE 2 Biosynthesis Among Individuals with Colorectal Adenomas: A Randomized Clinical Trial. Cancer Prev. Res. 2020.
- Fang, H.Y.; Lin, T.S.; Lin, J.P.;Wu, Y.C.; Chow, K.C.;Wang, L.S. Cyclooxygenase-2 in human non-small cell lung cancer. Eur. J. Surg. Oncol. 2003, 29, 171–177.
- Yoshimatsu, K.; Subbaramaiah, K.; Dannenberg, A.J.; DeLellis, R.A.; Golijanin, D.; Paty, P.B.; Soslow, R.A.; Jakobsson, P.J. Inducible microsomal prostaglandin E synthase is overexpressed in colorectal adenomas and cancer. Clin. Cancer Res. 2001, 7, 3971–3976.
- Sano, H.; Kawahito, Y.;Wilder, R.L.; Hashiramoto, A.; Mukai, S.; Asai, K.; Kimura, S.; Kato, H.; Kondo, M.; Hla, T. Expression of Cyclooxygenase-1and -2 in Human Colorectal Cancer1. Cancer Res. 1995, 55, 3785–790.
- Roelofs, H.M.J.; Morsche, R.H.M.t.; van Heumen, B.W.H.; Nagengast, F.M.; Peters, W.H.M. Over-expression of COX-2 mRNA in colorectal cancer. BMC Gastroenterol. 2014, 14, 3–8.
- Half, E.; Tang, X.M.; Gwyn, K.; Sahin, A.; Wathen, K.; Sinicrope, F.A. Cyclooxygenase-2 expression in human breast cancers and adjacent ductal carcinoma in situ. Cancer Res. 2002, 62, 1676–1681.
- Mehrotra, S.; Morimiya, A.; Agarwal, B.; Konger, R.; Badve, S. Microsomal prostaglandin E2 synthase-1 in breast cancer: A potential target for therapy. J. Pathol. 2006, 208, 356–363.
- Khor, L.Y.; Bae, K.; Pollack, A.; Hammond, M.E.H.; Grignon, D.J.; Venkatesan, V.M.; Rosenthal, S.A.; Ritter, M.A.; Sandler, H.M.; Hanks, G.E.; et al. COX-2 expression predicts prostate-cancer outcome: Analysis of data from the RTOG 92-02 trial. Lancet Oncol. 2007, 8, 912–920.
- Finetti, F.; Terzuoli, E.; Giachetti, A.; Santi, R.; Villari, D.; Hanaka, H.; Radmark, O.; Ziche, M.; Donnini, S. MPGES-1 in prostate cancer controls stemness and amplifies epidermal growth factor receptor-driven oncogenicity. Endocr. Relat. Cancer 2015, 22.
- Panza, E.; de Cicco, P.; Ercolano, G.; Armogida, C.; Scognamiglio, G.; Anniciello, A.M.; Botti, G.; Cirino, G.; Ianaro, A. Di erential expression of cyclooxygenase-2 in metastatic melanoma a ects progression free survival. Oncotarget 2016, 7, 57077–57085.
- Koga, H.; Sakisaka, S.; Ohishi, M.; Kawaguchi, T.; Taniguchi, E.; Sasatomi, K.; Harada, M.; Kusaba, T.; Tanaka, M.; Kimura, R.; et al. Expression of cyclooxygenase-2 in human hepatocellular carcinoma: Relevance to tumor dedi erentiation. Hepatology 1999, 29, 688–696.
- Zang, S.; Ni, M.; Lian, Y.; Zhang, Y.; Liu, J.; Huang, A. Expression of microsomal prostaglandin E2 synthase-1 and its role in human hepatocellular carcinoma. Hum. Pathol. 2013, 44, 1681–1687.
- Zarghi, A.; Arfaei, S. Selective COX-2 Inhibitors: A Review of Their Structure-Activity Relationships. Iran. J. Pharm. Res. 2011, 10, 655–683.
- Harris, R. Beebe, Alshafie, Reduction in cancer risk by selective and nonselective cyclooxygenase-2 (COX-2) inhibitors. J. Exp. Pharmacol. 2012, 91.
- Hansen-Petrik, M.B.; McEntee, M.F.; Jull, B.; Shi, H.; Zemel, M.B.; Whelan, J. Prostaglandin E2 protects intestinal tumors fromnonsteroidal anti-inflammatory drug-induced regression in Apcmin/+ mice. Cancer Res. 2002, 62, 403–408.
- Yan, M.; Myung, S.J.; Fink, S.P.; Lawrence, E.; Lutterbaugh, J.; Yang, P.; Zhou, X.; Liu, D.; Rerko, R.M.; Willis, J.; et al.15-Hydroxyprostaglandin dehydrogenase inactivation as a mechanism of resistance to celecoxib chemoprevention of colon tumors. Proc. Natl. Acad. Sci. USA 2009, 106, 9409–9413.
- Myung, S.J.; Rerko, R.M.; Yan, M.; Platzer, P.; Guda, K.; Dotson, A.; Lawrence, E.; Dannenberg, A.J.; Lovgren, A.K.; Luo, G.; et al. 15-Hydroxyprostaglandin dehydrogenase is an in vivo suppressor of colon tumorigenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 12098–12102.
- Kawamori, T.; Uchiya, N.; Sugimura, T.; Wakabayashi, K. Enhancement of colon carcinogenesis by prostaglandin E2 administration. Carcinogenesis 2003, 24, 985–990.
- Rohwer, N.; Kühl, A.A.; Schebb, N.H.; Zopf, D.; Mcdonald, F.M.;Weylandt, K.-H. E ects of chronic low-dose aspirin treatment on tumor prevention in three mouse models of intestinal tumorigenesis. Cancer Med. 2020.
- Nakanishi, M.; Montrose, D.C.; Clark, P.; Nambiar, P.R.; Belinsky, G.S.; Cla ey, K.P.; Xu, D.; Rosenberg, D.W. Genetic deletion of mPGES-1 suppresses intestinal tumorigenesis. Cancer Res. 2008, 68, 3251–3259.
- Nakanishi, M.; Menoret, A.; Tanaka, T.; Miyamoto, S.; Montrose, D.C.; Vella, A.T.; Rosenberg, D.W. Selective PGE 2 suppression inhibits colon carcinogenesis and modifies local mucosal immunity. Cancer Prev. Res. 2011, 4, 1198–1208.
- Sasaki, Y.; Nakatani, Y.; Hara, S. Role of microsomal prostaglandin E synthase-1 (mPGES-1)-derived prostaglandin E2 in colon carcinogenesis, Prostaglandins Other Lipid Mediat. Prostaglandins Other Lipid Mediat. 2015, 121, 42–45.
- Howe, L.R.; Subbaramaiah, K.; Kent, C.V.; Zhou, X.K.; Chang, S.H.; Hla, T.; Jakobsson, P.J.; Hudis, C.A.; Dannenberg, A.J. Genetic deletion of microsomal prostaglandin e synthase-1 suppresses mouse mammary tumor growth and angiogenesis. Prostaglandins Other Lipid Mediat. 2013, 106, 99–105.
- Finetti, F.; Terzuoli, E.; Bocci, E.; Coletta, I.; Polenzani, L.; Mangano, G.; Alisi, M.A.; Cazzolla, N.; Giachetti, A.; Ziche, M.; et al. Pharmacological inhibition of microsomal prostaglandin E synthase-1 suppresses Epidermal growth factor receptor-mediated tumor growth and angiogenesis. PLoS ONE 2012, 7, e40576.
- Bergqvist, F.; Morgenstern, R.; Jakobsson, P.J. A review on mPGES-1 inhibitors: From preclinical studies to clinical applications. Prostaglandins Other Lipid Mediat. 2020, 147, 106383.
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016.


