 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Silvia Gobbi | + 3735 word(s) | 3735 | 2020-11-27 07:22:26 | | | |
| 2 | Dean Liu | -1320 word(s) | 2415 | 2020-12-14 03:08:54 | | | | |
| 3 | Dean Liu | -1320 word(s) | 2415 | 2020-12-14 03:10:06 | | |
Video Upload Options
The current therapeutic approach for the treatment of hormone dependent breast cancer includes interference with estrogen receptors via either selective modulators or estrogens deprivation, by preventing their biosynthesis with aromatase inhibitors. Severe side effects and acquired resistance are drawbacks of both drug classes, and the efforts to overcome these issues still allow for research in this field to be animated. This review reports on recent findings that have opened new avenues for reconsidering the role of aromatase enzyme (and estrogen receptors) leading to the possibility of looking at well-known targets in a new perspective.
1. Introduction
Despite the increasingly advanced understanding of the molecular biology underlying the onset and progression of breast cancer (BC), this disease still represents the type of cancer that most frequently affects women and one of the leading causes of death among females worldwide[1]. Several risk factors, namely family history and obesity, but also a genetic predisposition due to mutations of tumor-suppressor genes, such as BRCA1 (breast cancer susceptibility gene 1) and BRCA2 (breast cancer susceptibility gene 2), responsible for DNA repair, have been related to higher BC incidence[2]. Notably, while observing an increase in diagnoses, a reduction in mortality can also be noticed, mainly due to early identification and fast therapeutic treatment of BC. However, the occurrence of resistance or disease recurrence renders a long-term therapy even more complex.
In a high percentage of cases, BC has proven to be estrogen dependent, since high levels of these hormones are needed for its growth and proliferation. Therefore, the development of an effective therapy for the treatment of this pathology over the years has been relying on the inhibition of estrogens effects. In this respect, human aromatase (HA), key enzyme for the biosynthesis of estrogens, and estrogen receptors (ERs) have long been thoroughly investigated as primary targets for endocrine therapy, and in particular, recent findings regarding the structure of HA have paved the way for new studies in this field. Indeed, research has now turned the spotlight back on HA, stimulating the interest in this old target from a different point of view. In this review, the latest emerged approaches are reported, which could shed new light on the role of HA in the modulation of estrogens level and lead to the design and development of new drug candidates acting with a different mechanism, allowing the overcoming of intrinsic drawbacks of currently used drugs.
2. Aromatase Enzyme
HA catalyzes the conversion of ASD, TST and 16α-hydroxytestosterone to E1, E2 and 17β,16α-estriol (E3), respectively (Figure 1). Its activity is highly specific, being the only enzyme in humans capable of catalyzing the aromatization of androgens to estrogens[13][14].
HA performs its function thanks to the electrons supplied by NADPH-cytochrome P450 reductase (CPR), a transmembrane protein formed by two cofactor domains: the flavin adenine dinucleotide (FAD) domain, which also contains the NADPH binding site, and the flavin mononucleotide (FMN) domain (Figure 3). In addition, a linker domain is situated between the FMN and FAD/NADPH domains. The transfer of electrons is considered to occur from NADPH to FAD and then to FMN. It was observed that CPR can take a “closed” conformation, where the FMN domain is close to FAD/NADPH domain favoring the internal transfer of electrons, or an “open” conformation, where the two cofactor domains are far away. The latter conformation seems to facilitate the intermolecular transfer of electrons from FMN to monooxygenase. The electronic activity of CPR is therefore regulated by the interconversion from the closed to the open conformation[15][16][17].
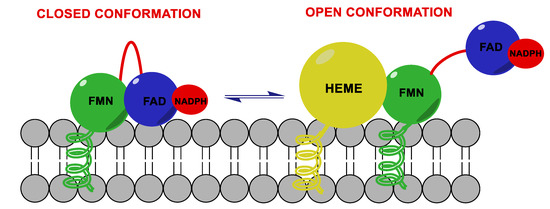
Figure 3. Aromatase complex. Flavin adenine dinucleotide (FAD) and flavin mononucleotide (FMN) domains of NADPH-cytochrome P450 reductase (CPR) can take a “closed” conformation, favoring the internal transfer of electrons, or an “open” conformation, where the two cofactor domains are far away, allowing the transfer of electrons from FMN to monooxygenase.
In 2009, Gosh et al. solved the crystallographic structure of the aromatase-ASD complex[18] for the first time, and this work allowed gaining new insight into the structure of this enzyme, until then hypothesized on the basis of sequence homology with other cytochromes P450[19][20]. HA is composed of a single polypeptide of 503 amino acids that fold up to form 12 α-helices (A–L) and 10 β-strands (1–10), which are distributed into four sheets, one major and three minor[21]. Considering the crystallographic structure, it was possible to establish that ASD binds to the steroid binding pocket directing its β-face towards the heme group and positioning its C19 4.0 Å from the Fe atom. This specific binding pocket at the active site is formed due to a 3.5 Å distortion of I-helix axis, this displacement being caused by Pro308, a specific residue of HA, absent in the structure of other cytochromes P450. This shift is stabilized by hydrogen bonds between Asp309 and Thr310 (2.8 Å) and between Asp309 and water (3.4 Å) and allows the Asp309 side chain to interact with 3-keto oxygen of ASD [18].
HA converts androgens to estrogens in a three-step process. The first step is a hydroxylation reaction on C19 methyl group producing a 19-hydroxy androgen, which is again hydroxylated in the second step forming a gem diol intermediate[22]. As for the last step, although several studies have been performed trying to understand the details of this reaction and postulating different routes[22][23][24], the underlying mechanism has not been fully elucidated yet.
3. Endocrine Therapy for Estrogen-Dependent Breast Cancer
To date, there are two main approaches for the treatment of estrogen positive (ER+) BC, directed at two different targets: the first relies on molecules that act on ERs by selectively modulating their activity, while the second focuses on blocking the endogenous synthesis of estrogens by inhibiting the aromatase enzyme.
4.1. Modulation of Estrogen Receptors
The first class of compounds developed for the treatment of ER + BC is represented by SERMs (selective estrogen receptors modulators), non-steroidal compounds binding to ERα and ERβ and endowed with a tissue-selective pharmacology, showing agonist activity in some tissues, such as bone, liver and cardiovascular system, and antagonist activity in other tissues, such as brain and breast, and mixed agonist/antagonist activity in the uterus[25][26]. This specificity is due to several factors, namely, tissue specific expression of the receptors, different availability of co-factors in various tissues and different conformational changes of the receptors induced by ligand binding[27]. Indeed, SERMs compete with estrogens and modulate ERs activity thanks to conformational changes that influence the binding of various cofactors, with which the receptors are associated. When estrogens bind to the hydrophobic pocket of the LBD, the receptor adopts a conformation in which helix 12 can seal the ligand into the ligand-binding pocket, activating AF2 and allowing the binding of cofactors to the receptor. Inversely, when a SERM binds to the LBD, its side chain prevents helix 12 from sealing the binding pocket and the repositioning of the helix hampers the binding of cofactors to AF2, blocking receptor activation[28][29]. The prototype of this class of compounds is tamoxifen (TAM, Figure 1), with a triphenylethylene structure, approved by the Food and Drug Administration (FDA) in 1977 and currently widely used for the treatment of ER + BC in pre- and postmenopausal women. TAM is administered as single Z-isomer (as the citrate salt), endowed with higher affinity for estrogens receptors than its E counterpart[30].
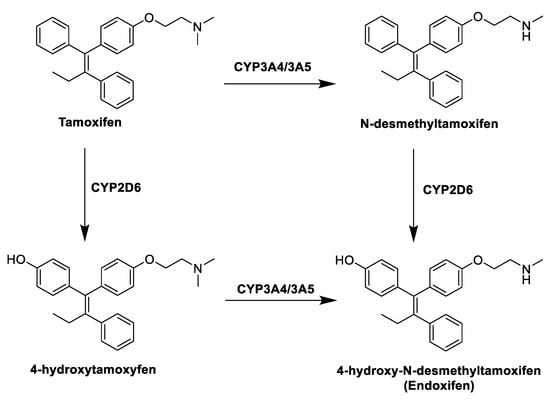
Figure 1. Tamoxifen (TAM) and its metabolites.
TAM is in fact a prodrug, and within the body it undergoes extensive metabolism by different cytochromes P450 that convert it into three active metabolites (Figure 1): 4-hydroxytamoxifen, N-desmethyltamoxifen and 4-hydroxydesmethyltamoxifen (endoxifen, END). The anticancer effect of TAM is thus mainly due to the activity of these metabolites, in particular 4-hydroxytamoxifen and END, produced by hydroxylation and demethylation of the drug by the action of hepatic CYP3A4/3A5 and CYP2D6[31]. Despite its therapeutic advantages, the use of TAM is limited by the development of intrinsic or acquired drug resistance[32] and its notable side effects, mostly in a long term therapy, among which the increased risk of developing endometrial cancer, caused by its agonist effect in the uterine tissue. This risk is dose- and time-dependent, and several studies have shown that patients taking TAM have two to three times greater risk of developing endometrial cancer than the rest of the population[33].
A more recent class of drugs that proved to be highly effective in modulating ERs is represented by selective estrogen receptor degraders (SERDs). The binding of these compounds to ER inhibits the activation of AF1 and AF2 domains, hinders the translocation of the receptor inside the nucleus and causes its degradation. Fulvestrant (Figure 2), a steroidal derivative, is the only SERD currently in use in BC therapy and is able to competitively bind the ER acting as pure antagonist. As it does not act as partial agonist in healthy tissues such as the uterus, its side effects are less pronounced than those of TAM[28][34].
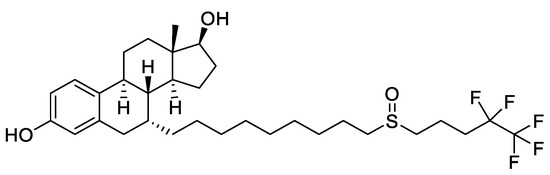
Figure 2. Structure of the selective estrogen receptor degrader (SERD) fulvestrant.
4.2. Aromatase Inhibitors
Due to its key role in the synthesis of estrogens, HA has long been regarded as a crucial target, to which small molecules could be directed in the development of endocrine therapy fighting BC. Aromatase inhibitors (AIs) bind to the enzyme and block its activity, inhibiting the endogenous synthesis of estrogens and drastically reducing the circulating levels of these hormones throughout the body. According to their chemical structure and their mechanisms of action, marketed AIs are divided into two classes, namely, steroidal and non-steroidal blockers. Steroidal AIs (exemestane, EXM[35], Figure 3) have a structure deriving from ASD, the natural substrate of HA, and they covalently bind the enzyme causing an irreversible inhibition. Non-steroidal AIs (anastrozole[36] and letrozole, LTZ[37], Figure 3) are derivatives featuring nitrogen-containing heterocycles that establish non-covalent interactions with the heme group of the enzyme by coordinating its Fe atom, resulting in a reversible inhibition[38][39][40][41].
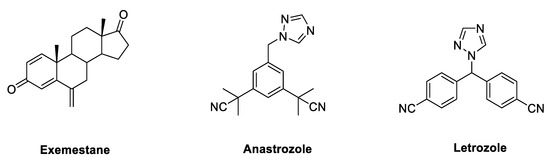
Figure 3. Marketed third generation aromatase inhibitors (AIs). These drugs feature either a steroidal (exemestane) or nonsteroidal (anastrozole and letrozole) structure.
These commercially available compounds, belonging to the third generation of AIs, have high specificity for HA without interfering with the biosynthesis of other steroid hormones and, to date, they are the first line endocrine therapy for the treatment of post-menopausal ER + BC. Nevertheless, the complete depletion of estrogen levels in the whole body caused by inhibition of this enzyme leads to the development of various side effects, such as musculoskeletal pain, reduction of bone density, increase of fractures and cardiovascular events [42][43][44][45]. Several studies have shown that third generation AIs, in particular LTZ, are superior to TAM as first-line therapy for advanced BC. In particular, the BIG (Breast International Group) 1-98 trial compared five years of TAM versus LTZ as monotherapy and the treatment for two years with one of these drugs followed by three years treatment with the other in postmenopausal women with ER-positive BC. It was found that treatment with LTZ as monotherapy led to an improvement in terms of disease-free survival, overall survival, distant recurrence-free interval and BC-free interval, with respect to TAM[46]. The ATAC (Arimidex, tamoxifen, alone or in combination) trial compared five years of anastrozole alone with TAM alone or in combination. Again, it was found that the treatment with AI led to an improvement of disease-free survival and time-to-recurrence compared with TAM. Moreover, treatment with anastrozole reduced the incidence of contralateral BC and of other drug-related adverse effects such as endometrial cancer, thromboembolic events, ischemic cerebrovascular events, hot flushes and vaginal discharge compared with TAM. On the other side, TAM led to a reduction of fractures and arthralgia observed with treatment with anastrozole[47][48]. For their superior clinical efficacy, it is reasonable to consider AIs as the adjuvant endocrine treatment of choice for post-menopausal women with this kind of cancer.
5. Emerging Roles for Aromatase Enzyme as BC Target
Despite the highly effective clinical outcome of the currently available therapies for ER+ BC, some issues still need to be fully addressed, and keep researchers’ attention focused on both the identification of novel targets involved in the onset of this disease and the optimization of the engagement of the validated ones. Indeed, the complete depletion of estrogens due to the treatment with AIs leads to increased frequency of cardiovascular events and, mainly, reduction of bone density. On the other side, SERMs also present significant side effects, especially in long-term therapies, among which is the increased risk of endometrial cancer. Moreover, the development of intrinsic or acquired drug resistance was also observed for both classes of drugs[32][49]. Thus, considerable efforts are devoted to the development of novel drugs that could help to overcome these issues, and a number of studies have also been carried out to get further insight into the specificities of the related targets. In particular, a significant crosstalk between HA and ERs was recently discovered, and the results of these studies opened new avenues for reconsidering the role of HA enzyme, leading to the potential development of novel classes of small molecules able to interfere in different modes with this well-known target.
The allosteric modulation of estrogen biosynthesis could offer significant advantages with respect to competitive drugs, since allosteric drugs could achieve the maximal inhibitory potency without a complete inhibition of estrogen production, and their effects would not be reduced at higher concentrations of natural substrates. Moreover, targeting allosteric sites, less conserved than active sites across protein families, may offer the possibility of developing more selective drugs, limiting off-target interactions and side effects.
On the other hand, the multitarget approach may lead to the identification of drugs endowed with an improved efficacy with respect to the widely used combination of drugs, overcoming the pharmacokinetic issues arising from the administration of separate compounds, simultaneously increasing patients’ compliance. The development of a single molecule able to reduce estrogens’ activity by interfering with different steps of the same pathway could also reduce the incidence of side effects usually seen with SERMs or AIs treatment. In this respect, recent studies seem to confirm the possibility of obtaining balanced AI/SERM dual target agents that could represent an effective strategy to tackle this disease.
In view of the results obtained in recent studies, the hybridation of molecules directed to different biological targets involved in the progression of ER+ BC could be further exploited by the use of a variety of agents (AIs, SERMs or SERDs) to obtain more effective drugs. Moreover, the latest studies on HA structure and the possibility of the fine tuning of its action via allosteric modulators have allowed for the reconsideration of this key enzyme from a new perspective, and the proof of principle of the feasibility of this approach will lead to a renewed interest for the search and development of new classes of AIs.
References
- Cokkinides, V.; Albano, J.; Samuels, A. American Cancer Society: Cancer Facts & Figures; American Cancer Society: Atlanta, GA, USA, 2020.
- Moynahan, M.E. The cancer connection: BRCA1 and BRCA2 tumor suppression in mice and humans. Oncogene 2002, 21, 8994–9007.
- Ghosh, D.; Lo, J.; Egbuta, C. Recent Progress in the Discovery of Next Generation Inhibitors of Aromatase from the Structure-Function Perspective. J. Med. Chem. 2016, 59, 5131–5148.
- Di Nardo, G.; Gilardi, G. Human aromatase: Perspectives in biochemistry and biotechnology. Biotechnol. Appl. Biochem. 2013, 60, 92–101.
- Ritacco, I.; Saltalamacchia, A.; Spinello, A.; Ippoliti, E.; Magistrato, A. All-Atom Simulations Disclose How Cytochrome Reductase Reshapes the Substrate Access/Egress Routes of Its Partner CYP450s. J. Phys. Chem. Lett. 2020, 11, 1189–1193.
- Iyanagi, T.; Xia, C.; Kim, J.J. NADPH-cytochrome P450 oxidoreductase: Prototypic member of the diflavin reductase family. Arch. Biochem. Biophys. 2012, 528, 72–89.
- Laursen, T.; Jensen, K.; Møller, B.L. Conformational changes of the NADPH-dependent cytochrome P450 reductase in the course of electron transfer to cytochromes P450. Biochim. Biophys. Acta 2011, 1814, 132–138.
- Ghosh, D.; Griswold, J.; Erman, M.; Pangborn, W. Structural basis for androgen specificity and oestrogen synthesis in human aromatase. Nature 2009, 457, 219–223.
- Favia, A.D.; Cavalli, A.; Masetti, M.; Carotti, A.; Recanatini, M. Three-dimensional model of the human aromatase enzyme and density functional parameterization of the iron-containing protoporphyrin IX for a molecular dynamics study of heme-cysteinato cytochromes. Proteins 2006, 62, 1074–1087.
- Karkola, S.; Höltje, H.D.; Wähälä, K. A three-dimensional model of CYP19 aromatase for structure-based drug design. J. Steroid Biochem. Mol. Biol. 2007, 105, 63–70.
- Ghosh, D.; Griswold, J.; Erman, M.; Pangborn, W. X-ray structure of human aromatase reveals an androgen-specific active site. J. Steroid Biochem. Mol. Biol. 2010, 118, 197–202.
- Akhtar, M.; Wright, J.N.; Lee-Robichaud, P. A review of mechanistic studies on aromatase (CYP19) and 17α-hydroxylase-17,20-lyase (CYP17). J. Steroid Biochem. Mol. Biol. 2011, 125, 2–12.
- Sgrignani, J.; Cavalli, A.; Colombo, G.; Magistrato, A. Enzymatic and Inhibition Mechanism of Human Aromatase (CYP19A1) Enzyme. A Computational Perspective from QM/MM and Classical Molecular Dynamics Simulations. Mini Rev. Med. Chem. 2016, 16, 1112–1124.
- Yoshimoto, F.K.; Guengerich, F.P. Mechanism of the third oxidative step in the conversion of androgens to estrogens by cytochrome P450 19A1 steroid aromatase. J. Am. Chem. Soc. 2014, 136, 15016–15025.
- Lewis-Wambi, J.S.; Jordan, V.C. Treatment of Postmenopausal Breast Cancer with Selective Estrogen Receptor Modulators (SERMs). Breast Dis. 2005, 24, 93–105.
- Wang, T.; You, Q.; Huang, F.S.; Xiang, H. Recent advances in selective estrogen receptor modulators for breast cancer. Mini Rev. Med. Chem. 2009, 9, 1191–1201.
- Martinkovich, S.; Shah, D.; Planey, S.L.; Arnott, J.A. Selective estrogen receptor modulators: Tissue specificity and clinical utility. Clin. Interv. Aging 2014, 9, 1437–1452.
- Patel, H.K.; Bihani, T. Selective estrogen receptor modulators (SERMs) and selective estrogen receptor degraders (SERDs) in cancer treatment. Pharmacol. Ther. 2018, 186, 1–24.
- Ring, A.; Dowsett, M. Mechanisms of tamoxifen resistance. Endocr. Relat. Cancer 2004, 11, 643–658.
- Osborne, C.K. Tamoxifen in the treatment of breast cancer. N. Engl. J. Med. 1998, 339, 1609–1618.
- Shagufta; Ahmad, I. Tamoxifen a pioneering drug: An update on the therapeutic potential of tamoxifen derivatives. Eur. J. Med. Chem. 2018, 143, 515–531.
- Rondón-Lagos, M.; Villegas, V.E.; Rangel, N.; Sánchez, M.C.; Zaphiropoulos, P.G. Tamoxifen Resistance: Emerging Molecular Targets. Int. J. Mol. Sci. 2016, 17, 1357.
- Burke, C. Endometrial cancer and tamoxifen. Clin. J. Oncol. Nurs. 2005, 9, 247–249.
- Blackburn, S.A.; Parks, R.M.; Cheung, K.L. Fulvestrant for the treatment of advanced breast cancer. Expert Rev. Anticancer 2018, 18, 619–628.
- Zucchini, G.; Geuna, E.; Milani, A.; Aversa, C.; Martinello, R.; Montemurro, F. Clinical utility of exemestane in the treatment of breast cancer. Int. J. Womens Health 2015, 7, 551–563.
- Kelly, C.M.; Buzdar, A.U. Anastrozole. Expert Opin. Drug Saf. 2010, 9, 995–1003.
- Dellapasqua, S.; Colleoni, M. Letrozole. Expert Opin. Drug Metab. Toxicol. 2010, 6, 251–259.
- Jiao, J.; Xiang, H.; Liao, Q. Recent advancement in nonsteroidal aromatase inhibitors for treatment of estrogen-dependent breast cancer. Curr. Med. Chem. 2010, 17, 3476–3487.
- Dutta, U.; Pant, K. Aromatase inhibitors: Past, present and future in breast cancer therapy. Med. Oncol. 2008, 25, 113–124.
- Chumsri, S.; Howes, T.; Bao, T.; Sabnis, G.; Brodie, A. Aromatase, aromatase inhibitors, and breast cancer. J. Steroid Biochem. Mol. Biol. 2011, 125, 13–22.
- Gobbi, S.; Rampa, A.; Belluti, F.; Bisi, A. Nonsteroidal aromatase inhibitors for the treatment of breast cancer: An update. Anticancer Agents Med. Chem. 2014, 14, 54–65.
- Khan, Q.J.; O’Dea, A.P.; Sharma, P. Musculoskeletal adverse events associated with adjuvant aromatase inhibitors. J. Oncol. 2010, 2010, 654348.
- Foglietta, J.; Inno, A.; de Iuliis, F.; Sini, V.; Duranti, S.; Turazza, M.; Tarantini, L.; Gori, S. Cardiotoxicity of Aromatase Inhibitors in Breast Cancer Patients. Clin. Breast Cancer 2017, 17, 11–17.
- Condorelli, R.; Vaz-Luis, I. Managing side effects in adjuvant endocrine therapy for breast cancer. Expert Rev. Anticancer 2018, 18, 1101–1112.
- Cepa, M.; Vaz, C. Management of bone loss in postmenopausal breast cancer patients treated with aromatase inhibitors. Acta Reum. Port. 2015, 40, 323–330.
- Ruhstaller, T.; Giobbie-Hurder, A.; Colleoni, M.; Jensen, M.B.; Ejlertsen, B.; de Azambuja, E.; Neven, P.; Láng, I.; Jakobsen, E.H.; Gladieff, L.; et al. Adjuvant Letrozole and Tamoxifen Alone or Sequentially for Postmenopausal Women with Hormone Receptor-Positive Breast Cancer: Long-Term Follow-up of the BIG 1-98 Trial. J. Clin. Oncol. 2019, 37, 105–114.
- Baum, M.; Buzdar, A.; Cuzick, J.; Forbes, J.; Houghton, J.; Howell, A.; Sahmoud, T. Anastrozole alone or in combination with tamoxifen versus tamoxifen alone for adjuvant treatment of postmenopausal women with early-stage breast cancer. Cancer 2003, 98, 1802–1810.
- Howell, A.; Cuzick, J.; Baum, M.; Buzdar, A.; Dowsett, M.; Forbes, J.F.; Hoctin-Boes, G.; Houghton, J.; Locker, G.Y.; Tobias, J.S.; et al. Results of the ATAC (Arimidex, Tamoxifen, Alone or in Combination) trial after completion of 5 years’ adjuvant treatment for breast cancer. Lancet 2005, 365, 60–62.
- Dutta, U.; Pant, K., Aromatase inhibitors: past, present and future in breast cancer therapy. Med Oncol 2008, 25 (2), 113-24.
- Chumsri, S.; Howes, T.; Bao, T.; Sabnis, G.; Brodie, A., Aromatase, aromatase inhibitors, and breast cancer. J Steroid Biochem Mol Biol 2011, 125 (1-2), 13-22.
- Gobbi, S.; Rampa, A.; Belluti, F.; Bisi, A., Nonsteroidal aromatase inhibitors for the treatment of breast cancer: an update. Anticancer Agents Med Chem 2014, 14 (1), 54-65.
- Khan, Q. J.; O'Dea, A. P.; Sharma, P., Musculoskeletal adverse events associated with adjuvant aromatase inhibitors. J Oncol 2010, 2010.
- Foglietta, J.; Inno, A.; de Iuliis, F.; Sini, V.; Duranti, S.; Turazza, M.; Tarantini, L.; Gori, S., Cardiotoxicity of Aromatase Inhibitors in Breast Cancer Patients. Clin Breast Cancer 2017, 17 (1), 11-17.
- Condorelli, R.; Vaz-Luis, I., Managing side effects in adjuvant endocrine therapy for breast cancer. Expert Rev Anticancer Ther 2018, 18 (11), 1101-1112.
- Cepa, M.; Vaz, C., Management of bone loss in postmenopausal breast cancer patients treated with aromatase inhibitors. Acta Reumatol Port 2015, 40 (4), 323-30.
- Ruhstaller, T.; Giobbie-Hurder, A.; Colleoni, M.; Jensen, M. B.; Ejlertsen, B.; de Azambuja, E.; Neven, P.; Láng, I.; Jakobsen, E. H.; Gladieff, L.; Bonnefoi, H.; Harvey, V. J.; Spazzapan, S.; Tondini, C.; Del Mastro, L.; Veyret, C.; Simoncini, E.; Gianni, L.; Rochlitz, C.; Kralidis, E.; Zaman, K.; Jassem, J.; Piccart-Gebhart, M.; Di Leo, A.; Gelber, R. D.; Coates, A. S.; Goldhirsch, A.; Thürlimann, B.; Regan, M. M.; Group, m. o. t. B.-C. G. a. t. I. B. C. S., Adjuvant Letrozole and Tamoxifen Alone or Sequentially for Postmenopausal Women With Hormone Receptor-Positive Breast Cancer: Long-Term Follow-Up of the BIG 1-98 Trial. J Clin Oncol 2019, 37 (2), 105-114.
- Baum, M.; Buzdar, A.; Cuzick, J.; Forbes, J.; Houghton, J.; Howell, A.; Sahmoud, T.; ATAC (Arimidex, T. m. A. o. i. C. T. G., Anastrozole alone or in combination with tamoxifen versus tamoxifen alone for adjuvant treatment of postmenopausal women with early-stage breast cancer: results of the ATAC (Arimidex, Tamoxifen Alone or in Combination) trial efficacy and safety update analyses. Cancer 2003, 98 (9), 1802-10.
- Howell, A.; Cuzick, J.; Baum, M.; Buzdar, A.; Dowsett, M.; Forbes, J. F.; Hoctin-Boes, G.; Houghton, J.; Locker, G. Y.; Tobias, J. S.; Group, A. T., Results of the ATAC (Arimidex, Tamoxifen, Alone or in Combination) trial after completion of 5 years' adjuvant treatment for breast cancer. Lancet 2005, 365 (9453), 60-2.
- Hanamura, T.; Hayashi, S. I., Overcoming aromatase inhibitor resistance in breast cancer: possible mechanisms and clinical applications. Breast Cancer 2018, 25 (4), 379-391.


