2. Effects of PPARs in the Placenta and the Fetus
During pregnancy, the placental metabolism can adapt to the environment throughout pregnancy to adapt to the maternal nutritional status and meet the demands of the fetus
[31]. All three PPAR isoforms are expressed in the placenta
[26][32]. The PPARs promote placental developmental plasticity by regulating lipid, hormone, and glucose metabolic pathways, including lipidogenesis, steroidogenesis, glucose transporters, and placental signaling pathways
[33]. Although the role of each PPAR in placental function has not been fully determined, unique and common functions between these isoforms have been observed. Among the PPAR-isoforms, PPARγ appears to be a major regulator of the mammalian placenta
[34]. PPARγ was the first to be demonstrated in the placenta
[35]. In rodent placenta, PPARγ is largely expressed in the trophoblastic layer of the labyrinth zone
[36][37]. In human placenta, PPARγ is present in villous trophoblast and extravillous trophoblast
[38][39]. There is some evidence suggesting that PPARγ modulates villous trophoblast differentiation, oxidative pathways, inflammatory response, and barrier formation
[40][41]. Furthermore, dysregulation of both PPARα and PPARγ in the placenta has been implicated in common complications of pregnancy, such as gestational diabetes mellitus, intrauterine growth restriction, and preeclampsia
[42]. Their expression pattern is regulated at least partially by DNA methylation in the placenta, and the involvement of other PPAR-regulated processes, such as placenta-specific miRNAs, has just been discovered
[43]. Placental epigenetic regulation appears to provide a plausible connection between environmental exposures and fetal development. Studies have shown that changes in placental DNA methylation patterns have been associated with fetal growth after exposure to maternal risk conditions such as GDM, obesity, and preeclampsia
[44][45].
During the development of the human embryo and fetus, three isoforms are expressed in cells of the endoderm and mesoderm at early time points in gestation
[46]. PPARβ/δ was the first allotype to be expressed during embryonic development in rodents
[47]. PPARα and PPARγ are expressed first in the placenta and then in the fetus
[47][48]. The role of PPAR in development has been revealed by studies in PPAR knockout mice
[49]. The important role of PPARα in lipid catabolism in the fetal liver and heart is consistent with the function of PPARα in adult tissues
[50][51][52]. Knockout of PPARα in mice causes a high miscarriage rate, hepatic lipid accumulation, obesity, and prolonged inflammation
[53][54]. In the early stages of organogenesis in the rat embryo, only the PPARβ/δ isotype is expressed
[47] and plays an important role in the closure of the neural tube
[55]. The signaling pathway involved in PPARβ/δ activation associated with nervous system development is profoundly altered by maternal diabetes
[55]. PPARγ null mutations are lethal. The developmental defects in the placenta occur in parallel to developmental defects in the embryo
[56]. In fetuses from diabetic rats, the concentration of PPARγ endogenous is reduced
[57]. The capacity of PPARγ endogenous to prevent the overproduction of both NO and MMPs in fetuses from diabetic rats demonstrates its anti-inflammatory effects
[57]. PPARγ activation increases lipid concentrations in midgestation fetuses from diabetic rats
[58]. Collectively, these data indicate that PPARs-mediated mechanisms are involved in the fetal origins of health and disease.
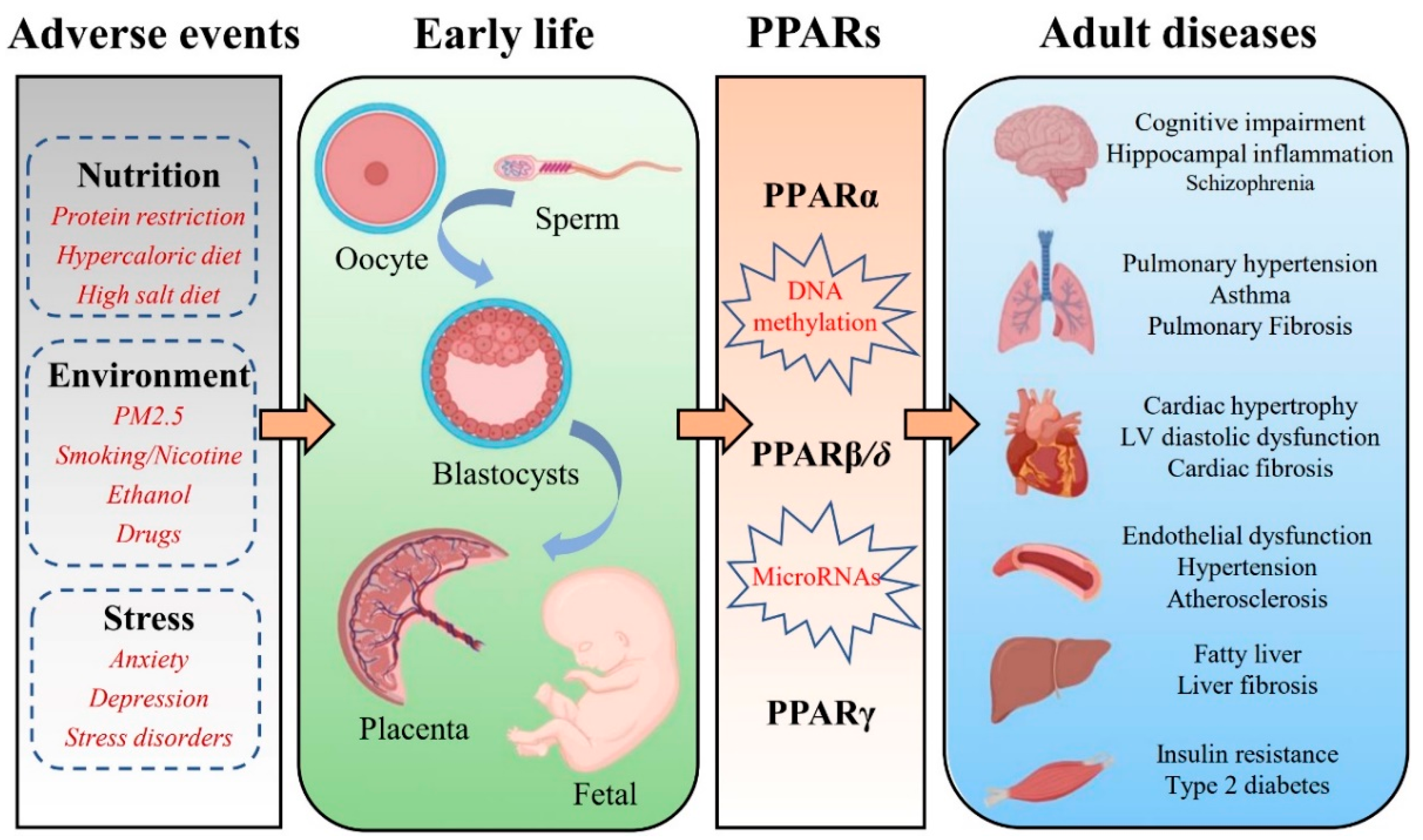
3. PPARs and FOAD
It is now well recognized that adverse events exposure in early life contribute to the development of the chronic diseases of adulthood, including hypertension, type 2 diabetes, stroke, cognitive impairment, and pulmonary hypertension. Additionally, the role of PPARs in numerous chronic diseases such as diabetes, cardiovascular diseases, autoimmune diseases, chronic fatigue, depression, and neurodegenerative diseases is well established. PPARs are ubiquitously expressed in almost all mammalian tissues and organs. Altering PPARs methylation patterns during early development may be maintained throughout the life course and even across generations
[31]. Researchers review the expression pattern of PPARs in various organs, including the brain, lung, heart, vessel, liver, and skeletal muscle, and discuss the potential roles of PPARs in FOAD (
Table 1).
Table 1. Summary of studies on PPARs in the fetal origins of adult disease.
| Organs |
Adverse Factors |
PPARs |
Adverse Outcomes/Phenotype |
Reference |
| Brain |
Maternal dietary restriction |
PPARα |
Abnormal sleep homeostatic regulation |
[59] |
| Maternal immune activation |
Disruption of dopamine function |
[60] |
| Maternal vitamin D deficiency |
PPARγ |
Angiogenesis impairment |
[61] |
| Maternal immune activation |
Cognitive impairments and anxiety behaviors |
[62] |
| Maternal high fructose |
Hippocampal neuroinflammation |
[63] |
| Intrauterine growth restriction |
Neurodevelopment and neurocognitive impairment |
[64] |
| Lung |
Perinatal nicotine exposure |
PPARγ |
Asthma |
[65] |
| Perinatal nicotine exposure |
Lung dysplasia |
[66] |
| Perinatal nicotine exposure |
Lung mitochondrial dysfunction |
[67] |
| Intrauterine growth restriction |
Impairment of lung development |
[68] |
| Heart |
Maternal protein restriction |
PPARα |
Dysregulation of lipid metabolism |
[69] |
| Maternal diabetes |
Fetal hypertrophic cardiomyopathy |
[51] |
| Maternal diabetes |
Cardiac oxidative stress |
[70] |
| Maternal obesity |
PPARγ |
Fetal cardiac dysfunction |
[71] |
| Maternal protein restriction |
Cardiac fibrosis |
[72] |
| Maternal nutrient restriction |
Myocardial lipid deposition |
[73] |
| Vessel |
Preeclampsia |
PPARβ/δ |
Endothelial dysfunction |
[74] |
| Maternal protein restriction |
PPARγ |
Aortic remodeling |
[75] |
| Liver |
Maternal exposure to PFOA |
PPARα |
Liver damage |
[76] |
| Maternal high-fat diet |
Non-alcoholic fatty liver disease |
[77] |
| Maternal nicotine exposure |
Metabolic-associated fatty liver disease |
[78] |
| Unbalanced folates/vitamin B12 diet |
Lipid metabolism impairment |
[79] |
| Maternal high-fat diet |
Obesity |
[80] |
| Liver |
Maternal ethanol exposure |
PPARα |
Non-alcoholic fatty liver disease |
[81] |
| Paternal hyperglycemia |
Hepatic steatosis |
[82] |
| Maternal high-fat feeding |
PPARγ |
Metabolic dysfunction |
[83] |
| Maternal bisphenol A exposure |
Non-alcoholic fatty liver disease |
[84] |
| Skeletal muscle |
Maternal protein restriction |
PPARα |
Metabolic inflexibility |
[85] |
| Intrauterine growth retardation |
PPARβ/δ |
Insulin resistance |
[86] |
| Maternal/Paternal type 2 diabetes |
Insulin resistance |
[87] |
| Intrauterine growth retardation |
PPARγ |
Insulin resistance |
[88] |
| Maternal cafeteria diet |
Skeletal muscle development and metabolic disorders |
[89] |
3.1. Brain
PPARα is expressed in several regions of the central nervous system (CNS), and its specific biological function remains unclear
[90]. Various inflammatory parameters were significantly enhanced in PPARα KO mice
[91]. Neuroinflammation is considered a cause and/or contributing factor to neuronal degeneration
[92]. It suggests that PPARα attenuates the inflammatory response after ischemia/brain injury
[93]. Moreover, the activation of PPARα has anti-inflammatory properties and a beneficial impact on certain neurologic diseases, including Alzheimer’s disease (AD)
[94], Multiple sclerosis (MS)
[95], Huntington’s disease (HD)
[96], and Parkinson’s disease (PD)
[97]. Malnutrition during pregnancy affects sleep homeostasis and increases sleep pressure in offspring, which may be related to the increased PPARα mRNA expression in the hypothalamus
[59]. In a study by Felice et al., it was found that prenatal administration of fenofibrate (PPARα agonist) reduced the risk of schizophrenia-like behavior in male offspring of maternal immune activation (MIA) and emphasizes PPARα as a possible target for schizophrenia therapies
[60].
Although PPARβ/δ is the most abundant PPAR subtype in the CNS, its role is rarely studied
[90]. It has been suggested that the most important roles of PPARβ/δ in brain cells are antioxidant and anti-inflammatory effects
[98]. There also a study identified that the differentiation of neural and glial cells might be impacted by PPARβ/δ, which may also affect the metabolism of cholesterol in the brain
[99]. One study found that prenatal exposure to a high-fat diet increased the density of cells immunoreactive for PPARβ/δ in the hypothalamic paraventricular nucleus, perifornical lateral hypothalamus, and central nucleus of the amygdala
[100]. However, the clinical significance of this change and the potential role of PPARβ/δ in fetal origins of CNS diseases remains unclear.
PPARγ is the most studied subgroup of the PPAR family and has an important role in the CNS, including relieving endoplasmic reticulum stress and the inflammatory response
[101], the balance of cerebral metabolite
[102] and the maintenance of glucose homeostasis
[103]. Animal studies have demonstrated that maternal vitamin D deficiency leads to decreased PPARγ levels in the offspring’s brain and affects angiogenesis in the brain
[61]. Fetal hippocampal inflammation is significantly increased in immune-activated mothers, followed by cognitive deficits, which are highly correlated with hippocampal neurogenesis disorders in pre-pubertal male offspring. The PPARγ agonist pioglitazone improves neurogenesis, cognitive impairment, and anxious behavior in MIA offspring
[62]. Maternal high-fructose-induced hippocampal neuroinflammation in the adult female offspring. Adult female offspring exposed to high maternal fructose have decreased levels of PPARγ and endogenous antioxidant expression in the hippocampus, which leads to hippocampal neuroinflammation. An oral dose of pioglitazone (PPARγ agonist) effectively increases the expression of antioxidants and blocks neuroinflammation
[63]. Based on the findings described above, synthetic PPARγ agonists have been suggested as therapeutic medicines for the treatment of CNS diseases such as PD
[104], AD
[105], HD, and Autism spectrum disorder
[106].
3.2. Lung
PPARα has been implicated in the control of airway inflammation, but as yet, little is known about its role in lung disease. There is a mouse model of pulmonary fibrosis suggesting that PPARα regulates fibrosis
[107]. A study by Genovese et al. revealed that endogenous and exogenous PPARα ligands reduced bleomycin-induced lung injury in mice
[108]. Liu et al. found that the activity of PPARα was inhibited in lipopolysaccharide (LPS) induced acute lung injury (ALI)
[109]. By reducing oxidative stress and inflammation, which are both directly related to the activation of PPARα, eupatilin has a protective function in ALI
[110]. Taken together, they proposed that PPARα could be a potential therapeutic target for lung injury.
PPARβ/δ agonist inhibited the proliferation of lung fibroblasts and enhanced the antifibrotic properties of PPARγ agonist
[107]. The role of PPARβ/δ in pulmonary hypertension and lung cancer has received attention in recent years. According to epidemiological and experimental animal studies, prenatal hypoxia, intrauterine growth restriction (IUGR), and obesity raise the risk of pulmonary hypertension in offspring
[111]. Prostacyclin and prostacyclin mimetics are the cornerstone of treatment for patients with pulmonary arterial hypertension (PAH)
[112]. One study suggests that PPARβ/δ may be a potent target for prostacyclin mimics in the treatment of pulmonary hypertension. PPARβ/δ agonist (GW0742) mediates vascular relaxation and prevents the right heart from hypertrophy associated with pulmonary arterial hypertension
[113]. The role of PPARβ/δ in the negative growth regulation of lung cancer cells was first reported in an in vitro study
[114]. Using a variety of lung cancer models, one research group demonstrated that increased synthesis of the PPARβ/δ agonist (prostacyclin) inhibited lung tumorigenesis
[115]. These findings imply that PPARβ/δ may play a protective function in PAH and lung cancer.
PPARγ is expressed in many lung cells, including bronchial epithelial cells, airway smooth muscle (HASM) cells, fibroblasts, alveolar type II pneumocytes, and mononuclear phagocytes
[107][116]. The activation of PPARγ signaling is involved in the paracrine effect of interstitial fibroblasts and alveolar type II (ATII) cells, which is necessary to maintain alveolar homeostasis
[117]. The PPARγ gene depends on developmentally specific transcription of mRNA variants and epigenetics for normal tissue. Therefore, it is susceptible to epigenetic changes
[118]. There is evidence that perinatal damage, including exposure to nicotine or maternal tobacco smoke (MTS), IUGR, and preterm delivery, altered both epigenetic determinants and gene expression in the lung
[118]. It has been demonstrated that IUGR caused epigenetic modifications to the PPARγ gene in rat lungs
[119]. The levels of PPARγ mRNA variants, PPARγ protein, and downstream targets were decreased in the lung of neonatal rats
[68]. Numerous studies have shown an increase in asthma in offspring whose mothers smoked during pregnancy
[120]. Perinatal smoke/nicotine exposure is a recognized factor that affects lung growth and differentiation by down-regulating the expression of PPARγ
[121]. Downregulation of PPARγ causes lipid-rich alveolar mesenchymal fibroblasts to transdifferentiate into myofibroblasts, which is the cellular hallmark of chronic lung diseases such as asthma
[122][123]. PPARγ agonist (rosiglitazone) can effectively block asthma induced by perinatal smoke exposure
[67].
3.3. Heart
PPARs are the physiological master switches of the heart, which guide the energy metabolism of cardiomyocytes, thereby influencing pathological heart failure and diabetic cardiomyopathy
[124]. However, the roles of PPARs in heart function and the results of their respective agonists differ greatly in preclinical animal models and clinical studies
[125]. PPARα is highly expressed in the heart and can affect the expression of numerous genes implicated in the uptake and oxidation of cellular fatty acid (FA)
[126]. Therefore, it plays a major role in cardiac fatty acid homeostasis. Down-regulation of PPARα expression altered the expression of fatty acid-metabolizing proteins that lead to myocardial damage and fibrosis
[127]. The expression of fetal cardiac lipid metabolism genes (PPARα, fatty acid translocase, lipoprotein lipase, etc.) was reduced in offspring from mothers with high blood glucose levels, not accompanied by cardiac triglyceride deposition or cardiac hypertrophy
[51]. However, it has subsequently been suggested that the heart of adult offspring from diabetic rats showed increased lipid concentrations. The increased expression of PPARα in offspring from diabetic rats can prevent toxic lipid accumulation in the heart
[128]. There is also solid evidence that PPARα exerts a protective effect on cellular oxidative damage
[129]. Thus, chronic deactivation of the PPARα signaling pathway may disrupt the balance between oxidant production and antioxidant defenses and ultimately contribute to heart damage
[130]. In the 2-day-old and pre-pubertal stage progeny from diabetic rats, there was an increase in the expression of prooxidative/proinflammatory markers and PPARα protein expression in the hearts. Maternal treatment with mitochondrial antioxidants led to reductions in PPARα protein expression and pro-oxidant/ro-inflammatory markers and prevented the adverse programming of heart alterations in prepubertal offspring from diabetic rats
[70]. Both neonatal and adult hearts from the offspring of maternal protein restriction (PR) during pregnancy showed a reduction in the level of PPARα promoter methylation and an increase in PPARα mRNA expression
[69]. The possible implication of these findings is that the enhanced capacity of fatty acid β-oxidation leads to an increased risk of oxidative damage to offspring hearts.
PPARγ is expressed at very low levels in the adult heart
[131]. PPARγ activation in cardiomyocytes is associated with impaired cardiac function due to its lipogenic effect
[131]. Maternal obesity leads to cardiac hypertrophy, and left ventricular diastolic dysfunction in offspring might be related to persistent upregulation of PPARγ expression
[71]. In rat offspring programmed by the reduced protein diet during gestation, the PPARγ agonist (rosiglitazone) was shown to have beneficial effects by reducing cardiac fibrosis and enhancing myocardial vascularization
[72]. PPARγ activator therapy has a beneficial impact on risk factors for cardiovascular disease, but it also appears to have adverse effects on the cardiovascular system. It has been reported that treatment with rosiglitazone is associated with an increase in myocardial infarction (MI) or heart failure in humans
[132].
3.4. Vessel
Studies have shown that PPARs are present in all essential vascular cells, including monocyte-macrophages, endothelial cells, and vascular smooth muscle cells
[133]. PPARs influence lipid metabolism and vascular diseases such as atherosclerosis and hypertension
[134]. PPARα has been implicated in blood pressure regulation and vascular inflammation
[135]. PPARα was expressed in both vascular endothelial cells and vascular smooth muscle cells
[136]. Activation of PPARα blocks multiple pathways such as NF-κB and MAPK, which in turn inhibit the expression of many genes involved in vascular inflammation, oxidative stress, and cell growth and migration
[137]. In experimental hypertension models, PPARα ligands can reportedly lower blood pressure
[138]. PPARα was also associated with atherosclerotic processes
[139]. The administration of the fibrate class of PPARα agonists to patients with type 2 diabetes or dyslipidemia significantly slowed the development of atherosclerosis and reduced their risk of cardiovascular events
[140], but surprisingly, high-fat diet PPARα-null mice are more responsive to insulin, have lower blood pressures, and develop less atherosclerosis
[139].
Activation of PPARβ/δ has a significant effect on anti-hypertension
[141]. However, it is argued that PPARβ/δ agonist acts via interference with the ET-1 signaling and lower blood pressure through a PPARβ/δ-independent mechanism
[142]. Moreover, the reduction of vascular oxidative stress markers and improvement of endothelial dysfunction were observed after a high dose of the PPARβ/δ antagonist GSK0660
[143]. It has been shown that the offspring of rats with maternal diabetes have abnormal fetal programming of vascular endothelial function, which is linked to increased ER stress and may be attributed to the down-regulation of the AMPK/PPAR signaling cascade
[144].
Whether PPARγ is hypotensive or hypertensive is still under debate so far
[145]. Genetic studies showed impaired vascular smooth muscle contraction in response to alpha-adrenergic drugs and hypotension in a generalized PPARγ knockout mouse model
[146], which is very well in agreement with the findings by Tontonoz
[147]. These findings suggest that PPARγ has a hypertensive function in controlling blood pressure. However, activation of PPARγ has beneficial effects on hypertension in a number of animal and human studies
[148]. PPARγ activation may regulate blood pressure via modulating endothelial vasoactive factors such as prostacyclin, nitric oxide, and endothelin-1. Additionally, PPARγ may also be involved in vessel tone regulation by down-regulating ANG II receptor 1 (AT1-R) in vascular smooth muscle cells
[149]. Angiotensin II-induced endothelial dysfunction in adult offspring of pregnancy complicated with hypertension is associated with impaired endothelial PPARγ
[74]. Rosiglitazone (a PPARγ agonist) reduced blood pressure and attenuated vascular remodeling in perinatal low-protein offspring rats
[75]. Chronic treatment with rosiglitazone has also been shown to prevent impaired nitric oxide synthase-dependent responses induced by prenatal alcohol exposure
[150]. Collectively, it is widely believed that activation of PPARγ can moderately lower blood pressure and plays a protective role in endothelial dysfunction, vascular inflammation, and other pathological processes that lead to atherosclerosis
[151].
 +1 credit
+1 credit