 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Julia T. Stadler | + 6157 word(s) | 6157 | 2020-12-01 03:21:04 | | | |
| 2 | Rita Xu | -3367 word(s) | 2790 | 2020-12-11 10:13:32 | | |
Video Upload Options
In obese individuals, atherogenic dyslipidemia is a very common and important factor in the increased risk of cardiovascular disease. Adiposity-associated dyslipidemia is characterized by low high-density lipoprotein cholesterol (HDL-C) levels and an increase in triglyceride-rich lipoproteins. Several factors and mechanisms are involved in lowering HDL-C levels in the obese state and HDL quantity and quality is closely related to levels of adiponectin. Recent studies have shown that obesity profoundly alters HDL metabolism, resulting in altered HDL subclass distribution, composition, and function.
1. Introduction
The increasing prevalence of obesity in the last decades has become a major health problem worldwide. In Northern America and Europe, in particular, the number of overweight and obese people is ever increasing and is becoming more common in children and adolescents [1]. The causes of obesity are multifactorial, with the most important factors being excess calorie intake and lack of physical activity. Excessive body weight increases the risk of disease development, such as coronary artery disease, hypertension, type-2 diabetes mellitus, and dyslipidemia [2][3][4][5][6]. High levels of triglyceride-rich lipoproteins and low levels of high-density lipoprotein cholesterol (HDL-C) commonly characterize dyslipidemia in obesity. In obesity, not only HDL levels are altered, but an altered HDL distribution pattern and abnormal HDL metabolism have also been observed, which often leads to dysfunction of the HDL particles [7][8][9]. Consequently, the focus has shifted from studying the quantity of HDL to studying the quality of HDL [10].
2. HDL Metabolism, Structure, and Composition
2.1. HDL Metabolism
The biogenesis of HDL starts in the liver and the intestine, where apolipoprotein (apo) A-I is synthesized (Figure 1). After secretion, lipid-poor apoA-I interacts with the integral cell membrane protein ATP-binding cassette transporter A1 (ABCA1), which is abundantly expressed by hepatocytes and enterocytes [11]. Through interaction, apoA-I acquires lipids from the cellular lipid pool, generating nascent HDL particles. Additional lipids and apolipoproteins are acquired, which are derived from hydrolysis of triglyceride-rich lipoproteins. This process partly explains the strong inverse relationship of HDL-C and triglyceride levels, often observed in obese subjects [12]. The acquired cholesterol of HDL is further esterified by lecithin-cholesterol-acyl transferase (LCAT), forming mature HDL particles [13]. The reaction takes place at the surface of HDL and requires apoA-I as an activator for LCAT [14]. The generated HDL-associated cholesteryl-esters are partially transferred to apoB-containing lipoproteins by cholesteryl-ester transfer protein (CETP), usually in exchange for triglycerides. Another pathway for clearance of cholesteryl-ester in HDL is the direct uptake by the liver via scavenger receptor class B type 1 (SR-BI) [15]. After interaction of SR-BI with large cholesterol-rich HDL, cholesteryl-esters and free cholesterol are internalized and cholesterol is removed through the bile, while apoA-I dissociates [16][17].
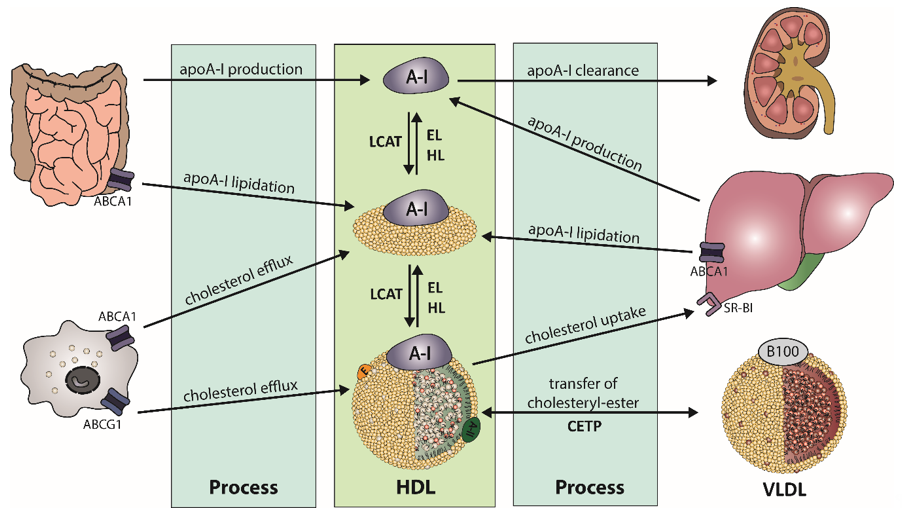
Figure 1. Schematic overview of high-density lipoprotein (HDL) metabolism. Biogenesis of apolipoprotein A-I (apoA-I) takes place in the liver and intestine. After secretion of the lipid-poor apoA-I, it interacts with ATP-binding cassette transporter A1 (ABCA1) to acquire lipids, leading to formation of nascent HDL. The enzyme lecithin-cholesterol-acyl transferase (LCAT) esterifies free cholesterol of nascent HDL to form mature HDL. Cholesteryl-esters are cleared by uptake of the liver by scavenger receptor B1 (SR-BI) or via transfer on triglyceride-rich lipoproteins by cholesteryl-ester transfer protein (CETP), in exchange of triglycerides. Triglyceride-rich HDL is susceptible to hydrolysis by endothelial lipase (EL) or hepatic lipase (HL).
HDL is enriched in triglycerides through the activity of CETP, generating HDL particles that are more susceptible to lipolysis by endothelial lipase (EL) or hepatic lipase (HL). Substrates for lipolysis are mainly phospholipids (EL) or phospholipids and triglycerides (HL), but with different specificity for phospholipids [18]. The lipolysis of triglycerides leads to the formation of smaller HDL particles, which are susceptible to faster catabolism. Another important key player of HDL metabolism is the phospholipid transfer protein (PLTP), which transfers phospholipids between HDL particles and lipids between triglyceride-rich lipoproteins and HDL [19]. Many apolipoproteins, lipid transfer proteins, enzymes, cell surface receptors, and cellular lipid transporters are involved in the regulation of HDL metabolism and partly determine levels of plasma HDL-C. This complex metabolism produces HDL particles of varying size, density, and composition. Therefore, plasma HDL-C concentrations are not a good parameter to reflect functional properties of HDL, such as HDL-mediated reverse cholesterol transport or anti-oxidative or anti-inflammatory properties.
2.2. HDL Structure and Composition
Plasma levels of HDL-C have been associated with cardiovascular diseases for decades [20][21][22]. However, it is becoming widely accepted that it is not the quantity but the quality of HDL that is important, as HDL performs different functions depending on the protein and lipid composition [23][24][25]. ApoA-I is the most prevalent protein component of HDL, accounting for approximately 70% of the total protein [26]. ApoA-I has a variety of functions, such as activation of LCAT, interaction with cellular receptors, and anti-atherogenic activities [27][28][29]. ApoA-II is the second major apolipoprotein in HDL and presents about 15–20% of the total protein component [30]. The remaining 10–15% of HDL protein mass comprises minor proteins, including apoA-IV, ApoCs, which are important enzyme regulators, apoD, apoE, apoF, apoH, apoJ, ApoL-I, and apoM, and several enzymes. Paraoxonase 1 (PON1) is almost exclusively associated with HDL and has been shown to exert anti-inflammatory and anti-oxidative properties [31]. Other enzymes associated with HDL are LCAT and the platelet-activating factor acetyl hydrolase. The phospholipid transfer protein and CETP have a lipid transfer activity and are important in lipoprotein metabolism. Remarkably, it is not cholesterol that predominates the HDL lipidome, but phospholipids. Taken together, phospholipids and sphingolipids account for 40–60% of total lipids, while cholesteryl-ester (30–40%), triglycerides (5–12%), and free cholesterol (5–10%) are less abundant [23]. Similar to functions of HDL-associated proteins, HDL lipids also accomplish distinct structural functions. The lipid surface monolayer is constituted of phospholipids, while cholesteryl-ester and triglycerides form the hydrophobic core. In total, more than 200 lipids and 80 proteins are carried by different HDL subclasses, with individual HDL particles carrying only a few other proteins besides apoA-I [32][33][34].
2.3. HDL Subclasses
Multiple subclasses of HDL exist, depending on its stage of maturation, site of origin, and its protein and lipid composition. Thus, HDL particles are highly heterogeneous in their size, shape, structure, and density (Table 1). Pre-β HDL is structurally the simplest form of HDL. These particles consist of one or two apoA-I molecules with a phospholipid layer and a trace amount of cholesterol. These particles are discoidal shaped with a diameter of approximately 9.6 nm and a thickness of 4.7 nm [35]. Pre-β HDL particles rapidly take up cholesterol and phospholipids, which convert them into larger HDL subclasses. Therefore, pre-β HDL only accounts for about 5% of HDL in the circulation [36]. Because of their function to avidly absorb cholesterol and phospholipids, pre-β HDL particles are thought to be a major factor in preventing atherosclerotic plaque formation. Importantly, higher serum cholesterol efflux capacity is related to plasma concentrations of pre-β HDL [37]. HDL3 particles have a smaller diameter (7.5 nm) and are enriched with proteins, while HDL2 particles are larger (10 nm) and lipid rich. Most abundant apolipoproteins are apoA-I and apoA-II in both subclasses; however, apoA-II is more present in HDL3. Interestingly, the HDL-associated enzyme PON1, which has anti-oxidative and anti-inflammatory properties [31], has been shown to be more frequently associated with HDL3. This higher abundance of PON1 on HDL3 could partly explain the higher anti-oxidative capacity of the smaller HDL particles [29][38]. HDL2 and HDL3 further show differences in lipid composition. Sphingolipids are, in general, less abundant in the HDL3 subclass, affecting surface lipid fluidity, whereas the bioactive lipid sphingosine-1-phosphate (S1P) is predominantly associated with HDL3 [23]. In line, the abundance of apoM, which specifically anchors S1P to HDL particle, shows higher abundance in HDL3 [38]. S1P maintains vascular integrity and mediates multiple effects of HDL on endothelial cells [39]. The functions of HDL to induce vasorelaxation as well as promoting barrier function have been attributed to signaling of S1P [40][41]. Taken together, it seems that smaller subclasses of HDL have a greater protective potential than larger particles [29].
Table 1. Representation of HDL heterogeneity.
|
HDL Subclass |
Size |
Shape |
Abundant Components |
Important Functions |
|
Pre-β HDL |
9.6 nm diameter, |
discoidal |
ApoA-I, phospholipids |
ABCA1-Cholesterol efflux |
|
HDL3 |
7.5 nm, 175 kDa |
spherical |
Protein:lipid ratio 55:45 PON1, ApoA-II, ApoM, S1P |
Anti-oxidative activity Anti-inflammatory activity ABCA1-Cholesterol efflux |
|
HDL2 |
10 nm, 350 kDa |
spherical |
Protein:lipid ratio 40:60 |
ABCG1- Cholesterol Efflux |
Apo, apolipoprotein; ABCA1, ATP-binding cassette transporter A1; PON1, paraoxonase 1; ABCG1, ATP-binding cassette subfamily G member 1.
2.4. Important Functions of HDL
One of the main functions of HDL is its ability to promote reverse cholesterol transport, the uptake of excess cholesterol from peripheral cells, and the transport to the liver for excretion. This process is considered as the major antiatherogenic effect of HDL [42].
The reverse cholesterol transport starts with the secretion of lipid-poor apoA-I, which is released from liver or intestine into the plasma to circulate to peripheral cells from which excess cholesterol is removed, forming nascent HDL. A key role in the reverse cholesterol transport is the interaction of apoA-I with ABCA1 [43]. Studies have shown that ABCA1 preferentially lipidates small HDL, specifically apoA-I, to form nascent HDL, while ATP-binding cassette subfamily G member 1 (ABCG1) stimulates cholesterol efflux to mature HDL and not to lipid-poor apoA-I [44][45]. Cholesterol efflux includes the passive diffusion of cholesterol from cells as well as the active cellular cholesterol transfer by ABCA1, ABCG1, and SR-BI [46][47][48]. The absorbed cholesterol is esterified by LCAT and mature HDL is formed. HDL-associated cholesteryl-ester is partially transferred to triglyceride-rich lipoproteins by CETP and further cleared by hepatic clearance through the low-density lipoprotein (LDL) receptor or taken up together with free cholesterol by the hepatic receptor SR-BI. Therefore, the transfer of cholesterol from peripheral cells to the liver involves two routes: (1) the direct uptake via SR-BI and (2) indirect by HDL-LDL/very low-density lipoprotein (VLDL) interaction [42]. In the liver, the cholesteryl-esters are hydrolyzed, and free cholesterol is either transported by ABCG5 and ABCG8 into the bile for excretion into feces or converted into bile acids or reused for VLDL production.
This process of HDL-mediated cholesterol efflux has been of expanded research interest in recent years. A number of different cell-based assays have been developed, to measure the ability of HDL to promote cholesterol efflux, the first step of reverse cholesterol transport. In the most established assay, a mouse macrophage cell line (J774) was employed [49]. Cells are enriched with radioactively or fluorescently labeled cholesterol and cyclic adenosine monophosphate to upregulate expression of ABCA1. For these assays, isolated HDL or apoB-depleted serum from patients is added to cell medium and the proportion between labeled cholesterol in the supernatant and in the cells is calculated.
Besides the ability of HDL to promote cholesterol efflux, there is increasing evidence that HDL-mediated antiatherogenic actions toward the endothelium have physiological relevance [50][51][52][53].
The beneficial properties of HDL on the endothelium include vasodilatory activity, primarily through stimulation of nitric oxide (NO) release from endothelial cells [40][54], and also the production of prostacyclin [55][56]. The initial step for the activation of NO production involves binding of HDL to SR-BI on the endothelium. Subsequent intracellular events are mediated by endothelial protein kinase B and intracellular Ca2+ mobilization, increase in intracellular ceramide levels, and the phosphorylation of the endothelial NO-synthase, leading to NO release [40][57][58][59]. HDL reduces the activity of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase in the endothelium, which reduces the cellular production of superoxide, an inactivator of NO, thereby increasing NO bioavailability [60]. Vasodilatory actions of HDL further include cholesterol efflux of cholesterol and 7-oxysterols, mediated by ABCG1, which improves formation of active endothelial NO-synthase dimers, resulting in decreased production of reactive oxygen species [61].
Another anti-atherogenic function of HDL is its ant-oxidative activity by protecting LDL from oxidative damage induced by free radicals, thus reducing its atherogenicity. ApoA-I, the major protein component of HDL may play a central role in HDL-mediated anti-oxidative activity, by reduction of lipid hydroperoxides through methionine residues [62][63]. In addition, HDL-associated PON1 was shown to decrease lipid peroxidation of LDL and HDL through a specific cysteine residue [64]. Other apolipoprotein components and HDL-associated enzymes, such as apoA-II, apoE, apoJ, lipoprotein-associated phospholipase A2, and LCAT, may further contribute to the anti-oxidative properties [29][65][66]. HDL-associated lipophilic antioxidants such as tocopherols seem to make a small contribution to the antioxidant properties of HDL [67].
Additionally, to the number of anti-oxidative effects, HDL further possesses anti-inflammatory properties. In vitro experiments have shown that HDL inhibits transmigration of monocytes [68] and inhibits cytokine-induced expression of vascular cell adhesion molecule, intercellular cell adhesion molecule, and E-selectin expression [69][70]. By modulation of the nuclear factor κB and the peroxisome proliferator-activated receptor gamma, HDL further inhibits the production of pro-inflammatory cytokines [71]. Due to these capabilities, HDL reduces the recruitment of lymphocytes, monocytes, and basophils to the vascular endothelium, thereby decelerating downstream events of inflammatory response.
3. Obesity, HDL, and Cardiovascular Risk
Obesity is one of the major risk factors for cardiovascular disease, which is associated with atherogenic dyslipidemia. These alterations in plasma lipid and lipoprotein levels contribute to the manifestation of such a severe morbidity.
3.1. Obesity Leads to a Shift in HDL Subclass Distribution
As described above, plasma HDL-C levels do not adequately reflect protective functions of HDL and greater protective potential is attributed to the smaller, more dense HDL particles. Recent studies of Woudberg et al. assessed HDL subclass distribution in normal-weight and obese white and black South African women. In obese study participants, a shift from large HDL toward increased levels of intermediate and small HDL subclasses was seen, whereby the effect was more pronounced in white women [72]. In a 5.5-year follow-up study they showed that the shifts in HDL subclass distribution were related to increasing central adiposity, suggesting a link between body fat distribution and lipid metabolism [8]. Based on the observed changes in HDL subclass distribution in obese individuals, Woudberg et al. explored the effect of exercise training on HDL subfractions. Interestingly, 12 weeks of exercise intervention altered the distribution of small HDL in obese women [73]. In adolescents suffering from type 2 diabetes mellitus, Davidson et al. determined the risk factors associated with the depletion of large HDL particles and simultaneous accumulation of small particles [74]. The authors investigated the distribution of HDL subclasses of individuals who differed in body mass index and insulin sensitivity and found that obesity is the major risk factor linked to the altered HDL subclasses. An increased CETP-mediated transfer of triglycerides on HDL and the subsequent hydrolysis of triglyceride-enriched HDL by hepatic lipase appeared to be the mechanism underlying the shift of large HDL to small and dense HDL particles [74].
3.2. Obesity Affects HDL Function
It is known that HDL functionality is severely impaired in certain diseases and HDL may even have inflammatory or pro-atherogenic properties. This was clearly demonstrated in HDL from patients suffering from chronic kidney disease [75][76], diabetes [77], cardiovascular disease [78], liver disease [79], psoriasis [80], or even atopic dermatitis [81] and allergic rhinitis [82]. Obesity-associated complications, such as inflammation or diabetes, have been shown to render HDL dysfunctional. HDL isolated from type 2 diabetes patients did not reduce endothelial oxidant stress and did not improve endothelium-dependent vasodilatation when compared to HDL isolated from healthy subjects [83]. Vasodilatory activity of HDL has been shown to be inversely correlated with triglyceride content of HDL, which is elevated in obesity [84]. A reduction of the overall capacity of HDL to promote cholesterol efflux from fibroblasts in obese, compared to lean, normal-weight, subjects was reported [85]. Of particular interest, cholesterol efflux capacity appears to be significantly inversely correlated with the body mass index [86][87]. Since cholesterol efflux capacity is the main metric of HDL function and has strong inverse association with coronary artery disease [88][89][90], the reduction of efflux capacity in obesity may have a crucial impact on the development of cardiovascular disease.
3.3. Adiponectin and HDL
It has been well reported that plasma HDL-C concentrations show a strong correlation with levels of adiponectin, independent of body mass index, distribution of body fat, and insulin sensitivity [91][92][93][94][95]. Adiponectin is mainly secreted by adipocytes, shows anti-atherogenic properties, and modulates glucose metabolism [96][97]. Studies with mice overexpressing or lacking adiponectin as well as in vitro studies suggest a causal relationship with HDL-C levels. Adiponectin increases the production of apoA-I as well as hepatic ABCA1, which increases HDL-C levels (Figure 3) [98][99]. The enhanced expression of ABCA1 has been suggested by activation of liver X receptor alpha and peroxisome proliferator-activated receptor gamma [100][101][102]. Plasma levels of adiponectin show a negative correlation with fractional catabolic rate of apoA-I in individuals with metabolic syndrome and control subjects [103][103]. Besides ABCA1, adiponectin upregulates ABCG1 expression, increases cholesterol efflux capacity, and effciently promotes lipidation of apoA-I, leading to formation of nascent HDL [104].
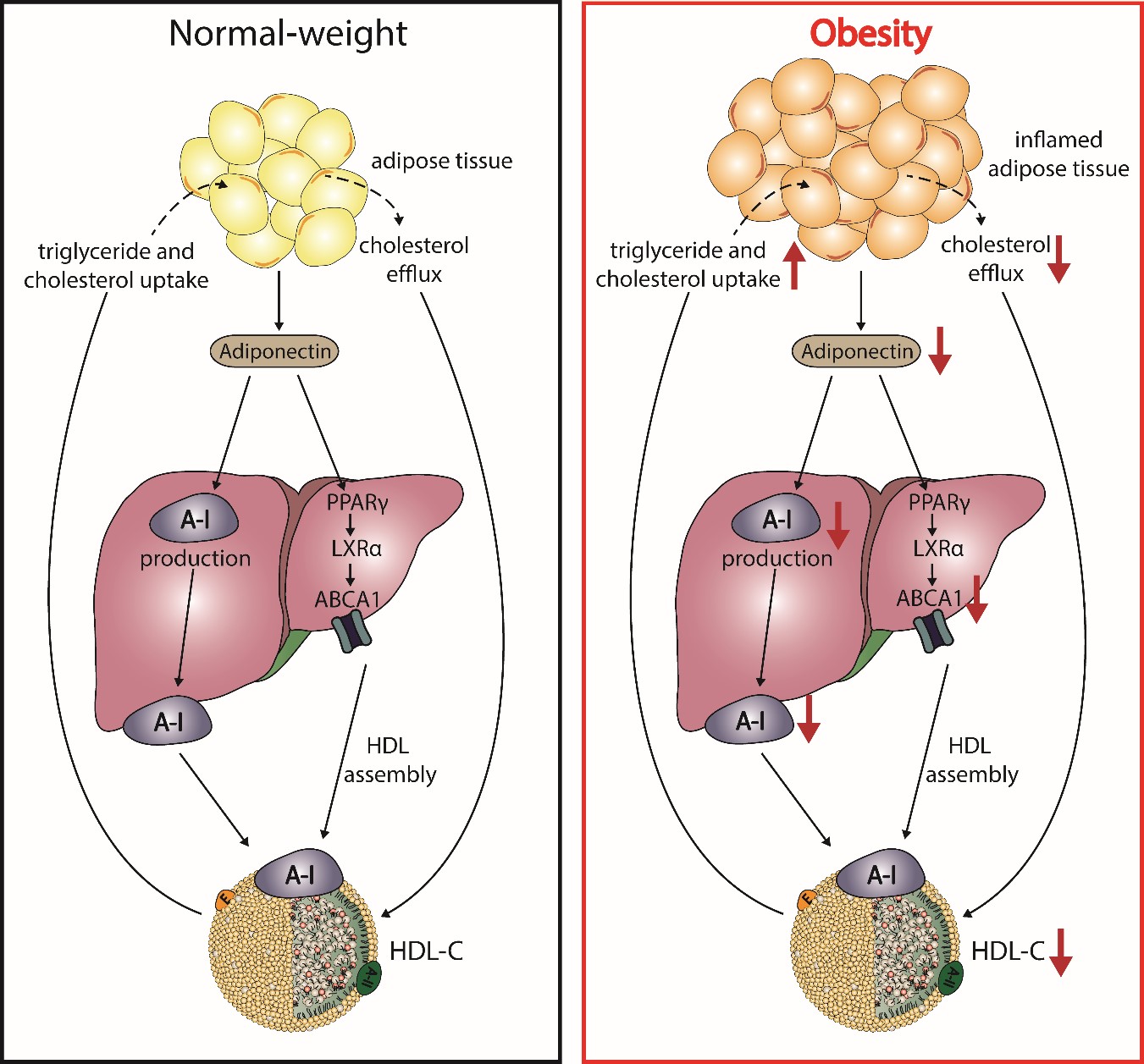
Figure 2. Postulated effects of obesity on adiponectin and HDL metabolism. In normal-weight subjects, adipocytes produce adiponectin, which enhances expression of the ATP-binding cassette transporter A1 (ABCA1) through activation of peroxisome proliferator-activated receptor gamma (PPARγ) and liver X receptor alpha (LXRα), leading to HDL assembly. Further, adiponectin increases the hepatic production of apoA-I. During the state of obesity, adipocytes manifest several altered properties, which play a role in the reduction of HDL-C. Increased inflammation and fat accumulation in the adipocytes reduces the production of adiponectin and impairs cholesterol flux to HDL. The reduction of adiponectin downregulates apoA-I production and ABCA1 expression in hepatocytes, thus reducing HDL assembly.
References
- Knight, J.A. Diseases and Disorders Associated with Excess Body Weight. Clin. Lab. Sci. 2011, 41, 107–121.
- Poirier, P.; Giles, T.D.; Bray, G.A.; Hong, Y.; Stern, J.S.; Pi-Sunyer, F.X.; Eckel, R.H. Obesity and cardiovascular disease: Pathophysiology, evaluation, and effect of weight loss. Thromb. Vasc. Biol. 2006, 26, 968–976, doi:10.1161/01.ATV.0000216787.85457.f3.
- Zeller, M.; Steg, P.G.; Ravisy, J.; Lorgis, L.; Laurent, Y.; Sicard, P.; Janin-Manificat, L.; Beer, J.C.; Makki, H.; Lagrost, A.C.; et al. Relation Between Body Mass Index, Waist Circumference, and Death After Acute Myocardial Infarction. Circulation 2008, 118, 482–490, doi:10.1161/CIRCULATIONAHA.107.753483.
- Al-Goblan, A.S.; Al-Alfi, M.A.; Khan, M.Z. Mechanism linking diabetes mellitus and obesity. Diabetes Metab. Syndr. Obes. Targets Ther. 2014, 7, 587–591, doi:10.2147/DMSO.S67400.
- Narkiewicz, K. Obesity and hypertension—The issue is more complex than we thought. Dial. Transplant. 2006, 21, 264–267, doi:10.1093/ndt/gfi290.
- Bays, H.E.; Toth, P.P.; Kris-Etherton, P.M.; Abate, N.; Aronne, L.J.; Brown, W.V.; Gonzalez-Campoy, J.M.; Jones, S.R.; Kumar, R.; La Forge, R.; et al. Obesity, adiposity, and dyslipidemia: A consensus statement from the National Lipid Association. Clin. Lipidol. 2013, 7, 304–383, doi:10.1016/j.jacl.2013.04.001.
- Rashid, S.; Genest, J. Effect of obesity on high-density lipoprotein metabolism. Obesity 2007, 15, 2875–2888, doi:10.1038/oby.2007.342.
- Woudberg, N.J.; Lecour, S.; Goedecke, J.H. HDL Subclass Distribution Shifts with Increasing Central Adiposity. Available online: https://www.hindawi.com/journals/jobe/2019/2107178/ (accessed on 10 August 2020).
- Wang, H.; Peng, D.-Q. New insights into the mechanism of low high-density lipoprotein cholesterol in obesity. Lipids Health Dis. 2011, 10, 176, doi:10.1186/1476-511X-10-176.
- Rader, D.J.; Tall, A.R. Is it time to revise the HDL cholesterol hypothesis? Med. 2012, 18, 1344–1346, doi:10.1038/nm.2937.
- Parks, J.S.; Chung, S.; Shelness, G.S. Hepatic ABC transporters and triglyceride metabolism. Opin. Lipidol. 2012, 23, 196–200, doi:10.1097/MOL.0b013e328352dd1a.
- Rader, D.J.; Hovingh, G.K. HDL and cardiovascular disease. Lancet 2014, 384, 618–625, doi:10.1016/S0140-6736(14)61217-4.
- Norum, K.R.; Remaley, A.T.; Miettinen, H.E.; Strøm, E.H.; Balbo, B.E.P.; Sampaio, C.A.T.L.; Wiig, I.; Kuivenhoven, J.A.; Calabresi, L.; Tesmer, J.J.; et al. Lecithin:cholesterol acyltransferase: Symposium on 50 years of biomedical research from its discovery to latest findings. Lipid Res. 2020, 61, 1142–1149, doi:10.1194/jlr.S120000720.
- Sorci-Thomas, M.G.; Bhat, S.; Thomas, M.J. Activation of lecithin: Cholesterol acyltransferase by HDL ApoA-I central helices. Lipidol. 2009, 4, 113–124, doi:10.2217/17584299.4.1.113.
- Acton, S.; Rigotti, A.; Landschulz, K.T.; Xu, S.; Hobbs, H.H.; Krieger, M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 1996, 271, 518–520, doi:10.1126/science.271.5248.518.
- Schaefer, E.J.; Anthanont, P.; Asztalos, B.F. HDL metabolism, composition, function and deficiency. Opin. Lipidol. 2014, 25, 194–199, doi:10.1097/MOL.0000000000000074.
- Kozarsky, K.F.; Donahee, M.H.; Rigotti, A.; Iqbal, S.N.; Edelman, E.R.; Krieger, M. Overexpression of the HDL receptor SR-BI alters plasma HDL and bile cholesterol levels. Nature 1997, 387, 414–417, doi:10.1038/387414a0.
- Duong, M.; Psaltis, M.; Rader, D.J.; Marchadier, D.; Barter, P.J.; Rye, K.-A. Evidence that hepatic lipase and endothelial lipase have different substrate specificities for high-density lipoprotein phospholipids. Biochemistry 2003, 42, 13778–13785, doi:10.1021/bi034990n.
- Albers, J.J.; Vuletic, S.; Cheung, M.C. Role of Plasma Phospholipid Transfer Protein in Lipid and Lipoprotein Metabolism. Biophys. Acta 2012, 1821, 345–357, doi:10.1016/j.bbalip.2011.06.013.
- Voight, B.F.; Peloso, G.M.; Orho-Melander, M.; Frikke-Schmidt, R.; Barbalic, M.; Jensen, M.K.; Hindy, G.; Hólm, H.; Ding, E.L.; Johnson, T.; et al. Plasma HDL cholesterol and risk of myocardial infarction: A mendelian randomisation study. Lancet 2012, 380, 572–580, doi:10.1016/S0140-6736(12)60312-2.
- Toth, P.P.; Barter, P.J.; Rosenson, R.S.; Boden, W.E.; Chapman, M.J.; Cuchel, M.; D’Agostino, R.B.; Davidson, M.H.; Davidson, W.S.; Heinecke, J.W.; et al. High-density lipoproteins: A consensus statement from the National Lipid Association. Clin. Lipidol. 2013, 7, 484–525, doi:10.1016/j.jacl.2013.08.001.
- Kannel, W.B.; Dawber, T.R.; Friedman, G.D.; Glennon, W.E.; Mcnamara, P.M. Risk Factors in coronary heart disease. An evaluation of several serum lipids as predictors of coronary heart disease; The Framingham Study. Intern. Med. 1964, 61, 888–899, doi:10.7326/0003-4819-61-5-888.
- Kontush, A.; Lhomme, M.; Chapman, M.J. Unraveling the complexities of the HDL lipidome. Lipid Res. 2013, 54, 2950–2963, doi:10.1194/jlr.R036095.
- Birner-Gruenberger, R.; Schittmayer, M.; Holzer, M.; Marsche, G. Understanding high-density lipoprotein function in disease: Recent advances in proteomics unravel the complexity of its composition and biology. Lipid Res. 2014, 56, 36–46, doi:10.1016/j.plipres.2014.07.003.
- Kontush, A.; Lindahl, M.; Lhomme, M.; Calabresi, L.; Chapman, M.J.; Davidson, W.S. Structure of HDL: Particle subclasses and molecular components. Exp. Pharmacol. 2015, 224, 3–51, doi:10.1007/978-3-319-09665-0_1.
- Kostner, G.; Alaupovic, P. Composition and structure of plasma lipoproteins. Separation and quantification of the lipoprotein families occurring in the high density lipoproteins of human plasma. Biochemistry 1972, 11, 3419–3428, doi:10.1021/bi00768a015.
- Wang, N.; Silver, D.L.; Costet, P.; Tall, A.R. Specific binding of ApoA-I, enhanced cholesterol efflux, and altered plasma membrane morphology in cells expressing ABC1. Biol. Chem. 2000, 275, 33053–33058, doi:10.1074/jbc.M005438200.
- Fernández-Hernando, C. Antiatherogenic Properties of High-Density Lipoprotein–Enriched MicroRNAs. Thromb. Vasc. Biol. 2014, 34, e13–e14, doi:10.1161/ATVBAHA.114.303542.
- Kontush, A.; Chapman, M.J. Antiatherogenic small, dense HDL--guardian angel of the arterial wall? Clin. Pract. Cardiovasc. Med. 2006, 3, 144–153, doi:10.1038/ncpcardio0500.
- Duriez, P.; Fruchart, J.C. High-density lipoprotein subclasses and apolipoprotein A-I. Chim. Acta Int. J. Clin. Chem. 1999, 286, 97–114, doi:10.1016/s0009-8981(99)00096-0.
- Litvinov, D.; Mahini, H.; Garelnabi, M. Antioxidant and Anti-Inflammatory Role of Paraoxonase 1: Implication in Arteriosclerosis Diseases. Am. J. Med. Sci. 2012, 4, 523–532, doi:10.4103/1947-2714.103310.
- Serna, J.; García-Seisdedos, D.; Alcázar, A.; Lasunción, M.Á.; Busto, R.; Pastor, Ó. Quantitative lipidomic analysis of plasma and plasma lipoproteins using MALDI-TOF mass spectrometry. Phys. Lipids 2015, 189, 7–18, doi:10.1016/j.chemphyslip.2015.05.005.
- Yetukuri, L.; Söderlund, S.; Koivuniemi, A.; Seppänen-Laakso, T.; Niemelä, P.S.; Hyvönen, M.; Taskinen, M.-R.; Vattulainen, I.; Jauhiainen, M.; Oresic, M. Composition and lipid spatial distribution of HDL particles in subjects with low and high HDL-cholesterol. Lipid Res. 2010, 51, 2341–2351, doi:10.1194/jlr.M006494.
- Wiesner, P.; Leidl, K.; Boettcher, A.; Schmitz, G.; Liebisch, G. Lipid profiling of FPLC-separated lipoprotein fractions by electrospray ionization tandem mass spectrometry. Lipid Res. 2009, 50, 574–585, doi:10.1194/jlr.D800028-JLR200.
- Jonas, A.; Kézdy, K.E.; Wald, J.H. Defined apolipoprotein A-I conformations in reconstituted high density lipoprotein discs. Biol. Chem. 1989, 264, 4818–4824.
- Woudberg, N.J.; Pedretti, S.; Lecour, S.; Schulz, R.; Vuilleumier, N.; James, R.W.; Frias, M.A. Pharmacological Intervention to Modulate HDL: What Do We Target? Pharmacol. 2018, 8, 989, doi:10.3389/fphar.2017.00989.
- De la Llera-Moya, M.; Drazul-Schrader, D.; Asztalos, B.F.; Cuchel, M.; Rader, D.J.; Rothblat, G.H. The Ability to Promote Efflux via ABCA1 Determines the Capacity of Serum Specimens with Similar HDL-C to Remove Cholesterol from Macrophages. Thromb. Vasc. Biol. 2010, 30, 796–801, doi:10.1161/ATVBAHA.109.199158.
- Davidson, W.S.; Silva, R.A.G.D.; Chantepie, S.; Lagor, W.R.; Chapman, M.J.; Kontush, A. Proteomic analysis of defined HDL subpopulations reveals particle-specific protein clusters: Relevance to antioxidative function. Thromb. Vasc. Biol. 2009, 29, 870–876, doi:10.1161/ATVBAHA.109.186031.
- Christoffersen, C.; Obinata, H.; Kumaraswamy, S.B.; Galvani, S.; Ahnström, J.; Sevvana, M.; Egerer-Sieber, C.; Muller, Y.A.; Hla, T.; Nielsen, L.B.; et al. Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Natl. Acad. Sci. USA 2011, 108, 9613–9618, doi:10.1073/pnas.1103187108.
- Nofer, J.R.; Van Der Giet, M.; Tölle, M.; Wolinska, I.; von Wnuck Lipinski, K.; Baba, H.A.; Tietge, U.J.; Gödecke, A.; Ishii, I.; Kleuser, B.; Schäfers, M. HDL induces NO-dependent vasorelaxation via the lysophospholipid receptor S1P3. Clin. Investig. 2004, 113, 569–581, doi:10.1172/JCI18004.
- Argraves, K.M.; Gazzolo, P.J.; Groh, E.M.; Wilkerson, B.A.; Matsuura, B.S.; Twal, W.O.; Hammad, S.M.; Argraves, W.S. High Density Lipoprotein-associated Sphingosine 1-Phosphate Promotes Endothelial Barrier Function. Biol. Chem. 2008, 283, 25074–25081, doi:10.1074/jbc.M801214200.
- Ouimet, M.; Barrett, T.J.; Fisher, E.A. HDL and Reverse Cholesterol Transport. Res. 2019, 124, 1505–1518, doi:10.1161/CIRCRESAHA.119.312617.
- Attie, A.D.; Kastelein, J.P.; Hayden, M.R. Pivotal role of ABCA1 in reverse cholesterol transport influencing HDL levels and susceptibility to atherosclerosis. Lipid Res. 2001, 42, 1717–1726.
- Tang, C.; Oram, J.F. The cell cholesterol exporter ABCA1 as a protector from cardiovascular disease and diabetes. Biophys. Acta BBA-Mol. Cell Biol. Lipids 2009, 1791, 563–572, doi:10.1016/j.bbalip.2009.03.011.
- Wang, N.; Lan, D.; Chen, W.; Matsuura, F.; Tall, A.R. ATP-binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high-density lipoproteins. Natl. Acad. Sci. USA 2004, 101, 9774–9779, doi:10.1073/pnas.0403506101.
- Kennedy, M.A.; Barrera, G.C.; Nakamura, K.; Baldán, Á.; Tarr, P.; Fishbein, M.C.; Frank, J.; Francone, O.L.; Edwards, P.A. ABCG1 has a critical role in mediating cholesterol efflux to HDL and preventing cellular lipid accumulation. Cell Metab. 2005, 1, 121–131, doi:10.1016/j.cmet.2005.01.002.
- Rothblat, G.H.; Phillips, M.C. High-density lipoprotein heterogeneity and function in reverse cholesterol transport. Opin. Lipidol. 2010, 21, 229.
- Phillips, M.C.; Johnson, W.J.; Rothblat, G.H. Mechanisms and Consequences of Cellular Cholesterol Exchange and Transfer. Available online: https://pubmed.ncbi.nlm.nih.gov/3297153/ (accessed on 14 October 2020).
- Marsche, G.; Heine, G.H.; Stadler, J.T.; Holzer, M. Current Understanding of the Relationship of HDL Composition, Structure and Function to Their Cardioprotective Properties in Chronic Kidney Disease. Biomolecules 2020, 10, 1348, doi:10.3390/biom10091348.
- Assmann, G.; Gotto, A.M. HDL Cholesterol and Protective Factors in Atherosclerosis. Circulation 2004, 109, III–8, doi:10.1161/01.CIR.0000131512.50667.46.
- Nofer, J.-R.; Assmann, G. Atheroprotective effects of high-density lipoprotein-associated lysosphingolipids. Trends Cardiovasc. Med. 2005, 15, 265–271, doi:10.1016/j.tcm.2005.08.005.
- Nofer, J.-R.; Kehrel, B.; Fobker, M.; Levkau, B.; Assmann, G.; von Eckardstein, A. HDL and arteriosclerosis: Beyond reverse cholesterol transport. Atherosclerosis 2002, 161, 1–16, doi:10.1016/s0021-9150(01)00651-7.
- Navab, M.; Ananthramaiah, G.M.; Reddy, S.T.; Van Lenten, B.J.; Ansell, B.J.; Fonarow, G.C.; Vahabzadeh, K.; Hama, S.; Hough, G.; Kamranpour, N.; et al. The oxidation hypothesis of atherogenesis: The role of oxidized phospholipids and HDL. Lipid Res. 2004, 45, 993–1007, doi:10.1194/jlr.R400001-JLR200.
- Mineo, C.; Deguchi, H.; Griffin, J.H.; Shaul, P.W. Endothelial and antithrombotic actions of HDL. Res. 2006, 98, 1352–1364, doi:10.1161/01.RES.0000225982.01988.93.
- Norata, G.D.; Callegari, E.; Inoue, H.; Catapano, A.L. HDL3 Induces Cyclooxygenase-2 Expression and Prostacyclin Release in Human Endothelial Cells Via a p38 MAPK/CRE-Dependent Pathway: Effects on COX-2/PGI-Synthase Coupling. Thromb. Vasc. Biol. 2004, 24, 871–877, doi:10.1161/01.ATV.zhq0504.1403.
- Beitz, J.; Förster, W. Influence of human low density and high density lipoprotein cholesterol on the in vitro prostaglandin I2 synthetase activity. Biophys. Acta BBA-Lipids Lipid Metab. 1980, 620, 352–355, doi:10.1016/0005-2760(80)90126-5.
- Drew, B.G.; Fidge, N.H.; Gallon-Beaumier, G.; Kemp, B.E.; Kingwell, B.A. High-density lipoprotein and apolipoprotein AI increase endothelial NO synthase activity by protein association and multisite phosphorylation. Natl. Acad. Sci. USA 2004, 101, 6999–7004, doi:10.1073/pnas.0306266101.
- Li, X.-A.; Titlow, W.B.; Jackson, B.A.; Giltiay, N.; Nikolova-Karakashian, M.; Uittenbogaard, A.; Smart, E.J. High density lipoprotein binding to scavenger receptor, Class B, type I activates endothelial nitric-oxide synthase in a ceramide-dependent manner. Biol. Chem. 2002, 277, 11058–11063, doi:10.1074/jbc.M110985200.
- Yuhanna, I.S.; Zhu, Y.; Cox, B.E.; Hahner, L.D.; Osborne-Lawrence, S.; Lu, P.; Marcel, Y.L.; Anderson, R.G.; Mendelsohn, M.E.; Hobbs, H.H.; et al. High-density lipoprotein binding to scavenger receptor-BI activates endothelial nitric oxide synthase. Med. 2001, 7, 853–857, doi:10.1038/89986.
- Van Linthout, S.; Spillmann, F.; Lorenz, M.; Meloni, M.; Jacobs, F.; Egorova, M.; Stangl, V.; De Geest, B.; Schultheiss, H.P.; Tschöpe, C. Vascular-Protective Effects of High-Density Lipoprotein Include the Downregulation of the Angiotensin II Type 1 Receptor. Hypertension 2009, 53, 682–687, doi:10.1161/HYPERTENSIONAHA.108.118919.
- Chen, W.; Xiao, H.; Rizzo, A.N.; Zhang, W.; Mai, Y.; Ye, M. Endothelial nitric oxide synthase dimerization is regulated by heat shock protein 90 rather than by phosphorylation. PLoS ONE 2014, 9, e105479, doi:10.1371/journal.pone.0105479.
- Panzenböck, U.; Stocker, R. Formation of methionine sulfoxide-containing specific forms of oxidized high-density lipoproteins. Biophys. Acta BBA-Proteins Proteom. 2005, 1703, 171–181, doi:10.1016/j.bbapap.2004.11.003.
- Garner, B.; Waldeck, A.R.; Witting, P.K.; Rye, K.A.; Stocker, R. Oxidation of high density lipoproteins. II. Evidence for direct reduction of lipid hydroperoxides by methionine residues of apolipoproteins AI and AII. Biol. Chem. 1998, 273, 6088–6095, doi:10.1074/jbc.273.11.6088.
- Aviram, M.; Billecke, S.; Sorenson, R.; Bisgaier, C.; Newton, R.; Rosenblat, M.; Erogul, J.; Hsu, C.; Dunlop, C.; La Du, B. Paraoxonase active site required for protection against LDL oxidation involves its free sulfhydryl group and is different from that required for its arylesterase/paraoxonase activities: Selective action of human paraoxonase allozymes Q and R. Thromb. Vasc. Biol. 1998, 18, 1617–1624, doi:10.1161/01.atv.18.10.1617.
- Miyata, M.; Smith, J.D. Apolipoprotein E allele-specific antioxidant activity and effects on cytotoxicity by oxidative insults and beta-amyloid peptides. Genet. 1996, 14, 55–61, doi:10.1038/ng0996-55.
- Kontush, A.; Chapman, M.J. Functionally defective high-density lipoprotein: A new therapeutic target at the crossroads of dyslipidemia, inflammation, and atherosclerosis. Rev. 2006, 58, 342–374, doi:10.1124/pr.58.3.1.
- Goulinet, S.; Chapman, M.J. Plasma LDL and HDL subspecies are heterogenous in particle content of tocopherols and oxygenated and hydrocarbon carotenoids. Relevance to oxidative resistance and atherogenesis. Thromb. Vasc. Biol. 1997, 17, 786–796, doi:10.1161/01.atv.17.4.786.
- Navab, M.; Imes, S.S.; Hama, S.Y.; Hough, G.P.; Ross, L.A.; Bork, R.W.; Valente, A.J.; Berliner, J.A.; Drinkwater, D.C.; Laks, H. Monocyte transmigration induced by modification of low density lipoprotein in cocultures of human aortic wall cells is due to induction of monocyte chemotactic protein 1 synthesis and is abolished by high density lipoprotein. Clin. Investig. 1991, 88, 2039–2046.
- Cockerill, G.W.; Rye, K.A.; Gamble, J.R.; Vadas, M.A.; Barter, P.J. High-density lipoproteins inhibit cytokine-induced expression of endothelial cell adhesion molecules. Thromb. Vasc. Biol. 1995, 15, 1987–1994, doi:10.1161/01.atv.15.11.1987.
- Calabresi, L.; Franceschini, G.; Sirtori, C.R.; De Palma, A.; Saresella, M.; Ferrante, P.; Taramelli, D. Inhibition of VCAM-1 expression in endothelial cells by reconstituted high density lipoproteins. Biophys. Res. Commun. 1997, 238, 61–65, doi:10.1006/bbrc.1997.7236.
- Bursill, C.A.; Castro, M.L.; Beattie, D.T.; Nakhla, S.; van der Vorst, E.; Heather, A.K.; Barter, P.J.; Rye, K.-A. High-density lipoproteins suppress chemokines and chemokine receptors in vitro and in vivo. Thromb. Vasc. Biol. 2010, 30, 1773–1778, doi:10.1161/ATVBAHA.110.211342.
- Woudberg, N.J.; Goedecke, J.H.; Blackhurst, D.; Frias, M.; James, R.; Opie, L.H.; Lecour, S. Associationbetween ethnicity and obesity with high-density lipoprotein (HDL) function and subclass distribution.Lipids Health Dis. 2016, 15, 92
- Woudberg, N.J.; Mendham, A.E.; Katz, A.A.; Goedecke, J.H.; Lecour, S. Exercise intervention alters HDLsubclass distribution and function in obese women. Lipids Health Dis. 2018, 17, 232
- Davidson,W.S.; Heink, A.; Sexmith, H.; Dolan, L.M.; Gordon, S.M.; Otvos, J.D.; Melchior, J.T.; Elder, D.A.;Khoury, J.; Geh, E.; et al. Obesity is associated with an altered HDL subspecies profile among adolescentswith metabolic disease. J. Lipid Res. 2017, 58, 1916–1923.
- Holzer, M.; Schilcher, G.; Curcic, S.; Trieb, M.; Ljubojevic, S.; Stojakovic, T.; Scharnagl, H.; Kopecky, C.M.;Rosenkranz, A.R.; Heinemann, A.; et al. Dialysis Modalities and HDL Composition and Function. J. Am.Soc. Nephrol. 2015, 26, 2267–2276.
- Holzer, M.; Birner-Gruenberger, R.; Stojakovic, T.; El-Gamal, D.; Binder, V.; Wadsack, C.; Heinemann, A.;Marsche, G. Uremia Alters HDL Composition and Function. J. Am. Soc. Nephrol. 2011, 22, 1631–1641.
- Farbstein, D.; Levy, A.P. HDL dysfunction in diabetes: Causes and possible treatments. Expert Rev.Cardiovasc. Ther. 2012, 10, 353–361
- Kosmas, C.E.; Martinez, I.; Sourlas, A.; Bouza, K.V.; Campos, F.N.; Torres, V.; Montan, P.D.; Guzman, E.High-density lipoprotein (HDL) functionality and its relevance to atherosclerotic cardiovascular disease.Drugs Context 2018, 7, 212525.
- Trieb, M.; Horvath, A.; Birner-Gruenberger, R.; Spindelboeck, W.; Stadlbauer, V.; Taschler, U.; Curcic, S.;Stauber, R.E.; Holzer, M.; Pasterk, L.; et al. Liver disease alters high-density lipoprotein composition,metabolism and function. Biochim. Biophys. Acta 2016, 1861, 630–638
- Holzer, M.;Wolf, P.; Curcic, S.; Birner-Gruenberger, R.;Weger,W.; Inzinger, M.; El-Gamal, D.;Wadsack, C.;Heinemann, A.; Marsche, G. Psoriasis alters HDL composition and cholesterol e_ux capacity. J. Lipid Res.2012, 53, 1618–1624
- Trieb, M.; Wolf, P.; Knuplez, E.; Weger, W.; Schuster, C.; Peinhaupt, M.; Holzer, M.; Trakaki, A.; Eichmann, T.;Lass, A.; et al. Abnormal composition and function of high-density lipoproteins in atopic dermatitis patients.Allergy 2019, 74, 398–402
- Trakaki, A.; Sturm, G.J.; Pregartner, G.; Scharnagl, H.; Eichmann, T.O.; Trieb, M.; Knuplez, E.; Holzer, M.;Stadler, J.T.; Heinemann, A.; et al. Allergic rhinitis is associated with complex alterations in high-densitylipoprotein composition and function. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 1280–1292
- Sorrentino, S.A.; Besler, C.; Rohrer, L.; Meyer, M.; Heinrich, K.; Bahlmann, F.H.; Mueller, M.; Horváth, T.;Doerries, C.; Heinemann, M.; et al. Endothelial-vasoprotective e_ects of high-density lipoprotein areimpaired in patients with type 2 diabetes mellitus but are improved after extended-release niacin therapy.Circulation 2010, 121, 110–122
- Perségol, L.; Vergès, B.; Foissac, M.; Gambert, P.; Duvillard, L. Inability of HDL from type 2 diabetic patientsto counteract the inhibitory e_ect of oxidised LDL on endothelium-dependent vasorelaxation. Diabetologia2006, 49, 1380–1386
- Sasahara, T.; Nestel, P.; Fidge, N.; Sviridov, D. Cholesterol transport between cells and high density lipoproteinsubfractions from obese and lean subjects. J. Lipid Res. 1998, 39, 544–554.
- Marsche, G.; Zelzer, S.; Meinitzer, A.; Kern, S.; Meissl, S.; Pregartner, G.;Weghuber, D.; Almer, G.; Mangge, H.Adiponectin Predicts High-Density Lipoprotein Cholesterol E_ux Capacity in Adults Irrespective of BodyMass Index and Fat Distribution. J. Clin. Endocrinol. Metab. 2017, 102, 4117–4123.
- Talbot, C.P.J.; Plat, J.; Joris, P.J.; Konings, M.; Kusters, Y.H.A.M.; Schalkwijk, C.G.; Ritsch, A.; Mensink, R.P.HDL cholesterol e_ux capacity and cholesteryl ester transfer are associated with body mass, but are notchanged by diet-induced weight loss: A randomized trial in abdominally obese men. Atherosclerosis 2018,274, 23–28
- Rohatgi, A.; Khera, A.; Berry, J.D.; Givens, E.G.; Ayers, C.R.; Wedin, K.E.; Neeland, I.J.; Yuhanna, I.S.;Rader, D.R.; de Lemos, J.A.; et al. HDL Cholesterol E_ux Capacity and Incident Cardiovascular Events.N. Engl. J. Med. 2014, 371, 2383–2393.
- Saleheen, D.; Scott, R.; Javad, S.; Zhao, W.; Rodrigues, A.; Picataggi, A.; Lukmanova, D.; Mucksavage, M.L.;Luben, R.; Billheimer, J.; et al. Association of HDL cholesterol e_ux capacity with incident coronary heartdisease events: A prospective case-control study. Lancet Diabetes Endocrinol. 2015, 3, 507–513.
- Khera, A.V.; Cuchel, M.; de la Llera-Moya, M.; Rodrigues, A.; Burke, M.F.; Jafri, K.; French, B.C.; Phillips, J.A.;Mucksavage, M.L.;Wilensky, R.L.; et al. Cholesterol E_ux Capacity, High-Density Lipoprotein Function,and Atherosclerosis. N. Engl. J. Med. 2011, 364, 127–135.
- Yamamoto, Y.; Hirose, H.; Saito, I.; Tomita, M.; Taniyama, M.; Matsubara, K.; Okazaki, Y.; Ishii, T.; Nishikai, K.;Saruta, T. Correlation of the adipocyte-derived protein adiponectin with insulin resistance index and serumhigh-density lipoprotein-cholesterol, independent of body mass index, in the Japanese population. Clin. Sci.Lond. Engl. 2002, 103, 137–142.
- Ryo, M.; Nakamura, T.; Kihara, S.; Kumada, M.; Shibazaki, S.; Takahashi, M.; Nagai, M.; Matsuzawa, Y.;Funahashi, T. Adiponectin as a biomarker of the metabolic syndrome. Circ. J. O_. J. Jpn. Circ. Soc. 2004, 68,975–981
- Sattar, N.; Wannamethee, G.; Sarwar, N.; Tchernova, J.; Cherry, L.; Wallace, A.; Danesh, J.; Whincup, P.Adiponectin and Coronary Heart Disease. Circulation 2006, 114, 623–629.
- . Cnop, M.; Havel, P.J.; Utzschneider, K.M.; Carr, D.B.; Sinha, M.K.; Boyko, E.J.; Retzla_, B.M.; Knopp, R.H.;Brunzell, J.D.; Kahn, S.E. Relationship of adiponectin to body fat distribution, insulin sensitivity and plasmalipoproteins: Evidence for independent roles of age and sex. Diabetologia 2003, 46, 459–469.
- Martin, L.J.; Woo, J.G.; Daniels, S.R.; Goodman, E.; Dolan, L.M. The relationships of adiponectin with insulinand lipids are strengthened with increasing adiposity. J. Clin. Endocrinol. Metab. 2005, 90, 4255–4259.
- Khera, A.V.; Cuchel, M.; de la Llera-Moya, M.; Rodrigues, A.; Burke, M.F.; Jafri, K.; French, B.C.; Phillips, J.A.;Mucksavage, M.L.;Wilensky, R.L.; et al. Cholesterol E_ux Capacity, High-Density Lipoprotein Function,and Atherosclerosis. N. Engl. J. Med. 2011, 364, 127–135
- Nigro, E.; Scudiero, O.; Monaco, M.L.; Palmieri, A.; Mazzarella, G.; Costagliola, C.; Bianco, A.; Daniele, A.New Insight into Adiponectin Role in Obesity and Obesity-Related Diseases. BioMed Res. Int. 2014, 2014.
- Matsuura, F.; Oku, H.; Koseki, M.; Sandoval, J.C.; Yuasa-Kawase, M.; Tsubakio-Yamamoto, K.; Masuda, D.;Maeda, N.; Tsujii, K.; Ishigami, M.; et al. Adiponectin accelerates reverse cholesterol transport by increasinghigh density lipoprotein assembly in the liver. Biochem. Biophys. Res. Commun. 2007, 358, 1091–1095.
- Oku, H.; Matsuura, F.; Koseki, M.; Sandoval, J.C.; Yuasa-Kawase, M.; Tsubakio-Yamamoto, K.; Masuda, D.;Maeda, N.; Ohama, T.; Ishigami, M.; et al. Adiponectin deficiency suppresses ABCA1 expression and ApoA-Isynthesis in the liver. FEBS Lett. 2007, 581, 5029–5033
- Tsubakio-Yamamoto, K.; Matsuura, F.; Koseki, M.; Oku, H.; Sandoval, J.C.; Inagaki, M.; Nakatani, K.;Nakaoka, H.; Kawase, R.; Yuasa-Kawase, M.; et al. Adiponectin prevents atherosclerosis by increasingcholesterol e_ux from macrophages. Biochem. Biophys. Res. Commun. 2008, 375, 390–394
- Liang, B.; Wang, X.; Guo, X.; Yang, Z.; Bai, R.; Liu, M.; Xiao, C.; Bian, Y. Adiponectin upregulates ABCA1expression through liver X receptor alpha signaling pathway in RAW 264.7 macrophages. Int. J. Clin.Exp. Pathol. 2015, 8, 450–457.
- Chawla, A.; Boisvert, W.A.; Lee, C.-H.; La_tte, B.A.; Barak, Y.; Joseph, S.B.; Liao, D.; Nagy, L.; Edwards, P.A.;Curtiss, L.K.; et al. A PPAR-LXR-ABCA1 Pathway in Macrophages Is Involved in Cholesterol E_ux andAtherogenesis. Mol. Cell 2001, 7, 161–171
- Vergès, B.; Petit, J.M.; Duvillard, L.; Dautin, G.; Florentin, E.; Galland, F.; Gambert, P. Adiponectin is animportant determinant of apoA-I catabolism. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1364–1369.
- Hafiane, A.; Daskalopoulou, S.S. Adiponectin’s mechanisms in high-density lipoprotein biogenesis andcholesterol e_ux. Metabolism 2020, 113, 154393

