 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mara Dierssen | + 4533 word(s) | 4533 | 2020-11-30 07:22:42 | | | |
| 2 | Rita Xu | -1505 word(s) | 3028 | 2020-12-11 06:41:02 | | |
Video Upload Options
The discovery in the last decade of unique astroglial features that include their role in synaptic plasticity and memory function has broadened and refurbished the conception of brain function in health and disease. Astrocytes are both necessary and sufficient for memory function, and contribute to the pathophysiology of cognitive and intellectual disability disorders such as Alzheimer’s disease, Fragile X syndrome (FXS), or Down syndrome (DS). We review some of the most relevant studies demonstrating that astrocytes are involved in the synaptic pathology of the two most common genetic forms of intellectual disability (FXS and DS).
1. Introduction
Neurodevelopmental disorders are multifaceted conditions characterised by different degrees of intellectual disability and impairment in communication (verbal and non-verbal) and motor skills, among others. Intellectual disability is defined by an overall intelligence quotient below average and deficits in adaptive behaviours, with an early onset during childhood [1][2]. These disorders arise from genetic alterations and/or environmental factors that influence how the brain develops and have short and long-term consequences on cognition, social interaction, and behaviour.
Traditionally, neurodevelopmental disorders such as Down syndrome (DS), Fragile X syndrome (FXS), or Rett syndrome have been considered as synaptopathies [3][4][5], characterised by neuronal alterations, including abnormalities in dendritic architecture [6][7], with changes in the complexity of dendritic arborisation [7], and in spine number [6][7][8][9][10][11][12], shape and length [7][10][13], reflecting more immature spines [10][14]. These changes are accompanied by impaired synaptogenesis [15][16][17], alterations in synaptic transmission and broad deficits in synaptic plasticity [18][19][20][21], and are thought to contribute to learning and memory deficits.
Synaptic plasticity and, specifically, changes in the strength of synaptic connectivity of neurons activated at the time of learning, are thought to be the basis of memory formation [22][23]. However, the alteration of the neuronal component does not completely explain the synaptic alterations or the behavioural and cognitive deficits observed in intellectual disability. This is possibly the reason why strategies focused on restoring neuronal dendritic abnormalities, impaired synaptic plasticity, and neurotransmitter imbalance, only achieve partial recovery of these deficits both in mouse models [9][24] and in humans [24][25][26]. This highlights the need to consider alternative mechanisms that may contribute to cognitive pathology.
Lately, the discovery of unique astroglial features that include their role in synaptic plasticity and memory function has broadened and refurbished the conception of brain function in health and disease. Recent reports underscoring the astrocyte's capability to modulate neuronal circuit activity and to potentiate synapses [27][28] suggest that astrocytes are both necessary [29][30][31] and sufficient [28] for memory function. Moreover, studies on intellectual disabilities have recently uncovered potential contributions of astrocytes to their pathophysiology [16][32][33][34]. Increasing body of evidence suggests that changes in astrocyte physiology and morphology might be involved in Alzheimer’s disease, FXS, or DS, among others [35][36][37][38][39]. In these pathological conditions, astrocytes modify their function and exhibit some common pathological features including an increase in the number and size of astrocytes together with increased expression of astroglial proteins such as S100 calcium-binding protein β (S100β) [16][40][41][42], a calcium binding protein coded by a HSA21 gene, and the glial fibrillary acidic protein GFAP [36][42][43][44], the main intermediate filament proteins of mature astrocytes. In fact, these pathophysiological changes in astrocytes have been linked with reduced neuronal activity [45], spine defects [14][32][34], and impaired memory performance [33][46][47][48].
2. Astrocyte Dysfunction in Neurodevelopmental Disorders
The involvement of astroglia in the pathophysiology of neurodevelopmental disorders is supported by their numerous misregulated glial genes [40][49]. The proteins encoded by these genes play important roles in the brain, including their involvement in cell cycle progression, neuronal differentiation, and neuronal damage repair. Along with these genetic alterations, astrocytes display aberrant morphology [42][50] and physiology [41] that have been directly shown to contribute not only to synaptic defects and changes in neuronal excitability [41] but also to neuronal survival [51][52].
It is frequent to detect different degrees of astrocyte reactivity (or astrogliosis) in many brain disorders [36][42][50]. The term astrogliosis refers not only to astrocyte numbers, but also to changes at the molecular, cellular, and functional level [53]. In general, astrogliosis involves morphological and physiological alterations, such as an increase in the number and size of the astrocytes, and changes in the expression of astroglial proteins (GFAP and S100β) [53]. Astrogliosis, along with peripheral macrophages and microglia, promotes an adaptive state that helps facing the origin of the brain insult (infection, haemorrhage, etc.) by phagocytizing external factors, eliminating toxic neuronal debris, and/or promoting neuronal survival [53][54][55]. However, in cognitive disorders such as DS, FXS, or Alzheimer’s disease, astrocytes are in a chronic “reactive state”, a continuous dysfunctional mode that can be maladaptive and contribute to the progression of neurodegeneration and brain dysfunction [56][57].
The changes in astroglial function vary depending on the severity of the brain lesion or the genetic alteration, and have repercussions on adjacent neurons. Given the role of astrocytes in the regulation of synaptic function, it is not surprising that changes in astrocyte activity, protein secretion, deregulation in gene expression, or modification in the astroglial membrane channel composition that occur in neurodevelopmental disorders might impair synaptic transmission and, therefore, memory function. In the next section, we review some of the most relevant studies demonstrating that astrocytes are involved in the synaptic pathology in DS and FXS, the two most common genetic forms of intellectual disability.
2.1. Astrocyte Pathology in Fragile X Syndrome (FXS)
FXS is a genetic condition caused by an expansion of the CGG triplet within the fragile X mental retardation 1 gene (FMR1) that leads to its transcriptional silencing. FMR1 is located in the X chromosome and encodes for the mRNA binding protein Fragile X Mental Retardation Protein 1 (FMRP) that regulates protein synthesis. The absence of FMRP interferes with brain development and contributes to FXS pathophysiology [58][59]. Several lines of evidence suggest that astroglia might also contribute to synaptic function impairment and memory deficits of individuals with FXS [16][32][33]. However, studies on astrocyte pathology in FXS are sparse and inconsistent: one group reports no astrogliosis seen in post-mortem brains of persons with FXS [60] while other describes a gliosis in the CA4 hippocampal region of two postmortem FXS brains [61].
One of the most commonly used mouse models for the study of FXS is the Fmr1 knock-out (KO) that recapitulates most of the neuronal alterations and the phenotypical traits of FXS [62]. This model lacks the expression of FMRP protein in neurons and astrocytes being thus an interesting tool to uncover the role of astrocyte in FXS. It is still unclear, however, whether FMRP protein has similar or different functions when expressed in neurons or in astrocytes. In FXS, the FMRP protein regulates metabotropic glutamate receptor 5 (mGluR5) expression in astrocytes, but not in neurons. In Fmr1 KO astrocytes, the absence of FMRP leads to misregulation of mGluR5 and reduced expression of glutamate transporter 1 (GLT-1) with subsequent decrease of glutamate uptake in astrocytes [63] (Figure 1). Increased mGluR5 signalling in neurons has been long proposed to account for the syndromic features and the cognitive deficits in FXS [64][65]. In fact, the acute and chronic pharmacological inhibition of mGluR5 in adult Fmr1 KO mice restores dendritic alterations including aberrant dendritic morphology, increases protein synthesis, and rescues memory deficits associated with FXS [66][67]. In fact, mGluR5 is upregulated in neurons [68] but downregulated in astrocytes [63]. Thus, the contribution of mGluR5 dysregulation to FXS pathophysiology is more complex than expected. mGluR5 activation has been associated with a form of synaptic depression, called mGluR5-mediated LTD resulting from the internalization of surface-expressed α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR), as a response to mGluR5 activation in neurons [69][70]. This is accompanied by reduced presynaptic release of glutamate [71] that, overall, leads to an exaggerated LTD in Fmr1 KO mice [72]. Conversely, mGluR5 downregulation in Fmr1 KO astrocytes has been associated with lower astroglial GLT-1 levels [63]. This might contribute to reduced glutamate reuptake and, therefore, to increased extracellular glutamate levels which subsequently might activate mGluR5 receptors in postsynaptic neurons, thus promoting mGluR5-mediated LTD.
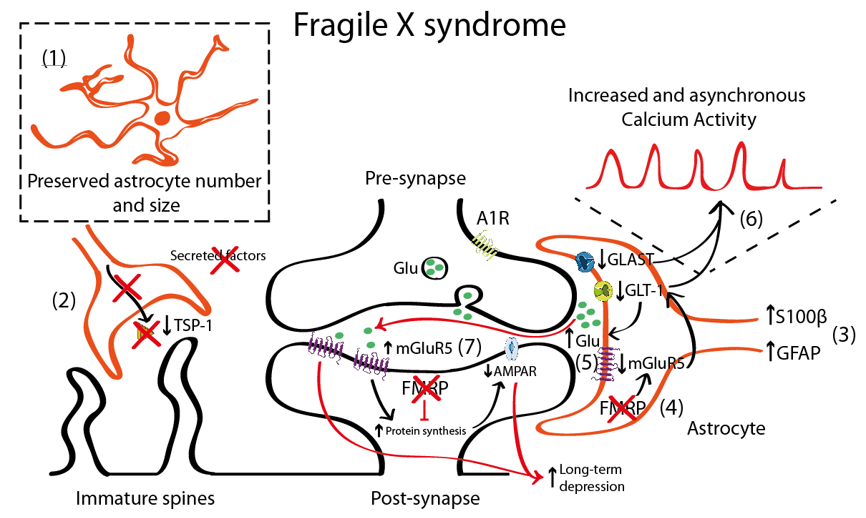
Figure 1. Schematic representation illustrating the astrocyte-synapse alterations in Fragile X syndrome (FXS). (1) Astrocyte number is preserved. (2) Reduced astrocyte secreted thrombospondin (TSP-1) prevents spine maturation resulting in more abundant filopodia (immature) spines. (3) Increased S100 calcium-binding protein β (S100β) and GFAP expression has been described in astrocytes. However, this altered expression has not been directly linked with their activity or function. (4) Fragile X Mental Retardation Protein (FMRP) absence in FXS astrocytes leads to metabotropic glutamate receptor 5 (mGluR5) downregulation in astrocytes (yet not in neurons) that negatively regulates glutamate transporter 1 (GLT-1) expression. Impaired glutamate transport due to decreased astrocyte glutamate-aspartate transporter 1 (GLAST-1) and GLT-1 expression increases extracellular glutamate levels (5) and astroglial calcium oscillations (6). This excess of glutamate might activate the postsynaptic mGluR5, which is overexpressed in neurons. (7) mGluR5 and FMRP oppositely regulate mRNA translation at the synapse: mGluR5 promotes it and FMRP prevents it. Therefore, increased mGluR5 expression and lack of FMRP in FXS leads to a disbalance in protein expression levels that account for many of the syndromic features of FXS including an α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) internalization that leads to an exaggerated mGluR5-mediated long-term depression (LTD) that is reported in Fmr1 knock-out (KO) mice.
Interestingly, increased GFAP expression, a marker of astrocyte activation, has been reported in the cortex, hippocampus, and striatum of Fmr1 KO mice [73], suggesting a reactive gliosis in these particular brain regions. In general, increased GFAP expression is accompanied by an increase in the number of astrocytes [53]. Nevertheless, there is only one description of persistent astrogliosis (increased GFAP and S100β expression) in the cerebellum of Fmr1 KO mice [74], but no systematic quantification of the astroglial number has been performed in FXS. Complementary to these data, astrocyte physiology was also reported to be altered in a mouse model with a gain-of-function of the premutation CGG (preCGG) repeat within the FMR1 gene. preCGG knock-in mice showed increased and asynchronous calcium activity that was explained by increased glutamate levels in the extracellular space due to reduced expression of glutamate transporters such as the glutamate-aspartate transporter 1 (GLAST-1) and GLT-1 in astrocytes [75].
In cellular FXS models, control hippocampal neurons grown in co-culture with FXS astrocytes, showed aberrant dendritic morphology and decreased expression of synaptic markers (PSD-95) [75]. Conversely, when co-cultured with control astrocytes, hippocampal FXS neurons developed normally. These dendritic alterations were explained by a reduction in the expression of thrombospondin (TSP-1) in Fmr1 KO mice astrocytes [32]. TSP-1 is synthesized and secreted by astrocytes [76], promoting synaptogenesis [77] and neurite outgrowth [78] both during the neurodevelopment [79] and in the adult brain [80]. Interestingly, both culturing Fmr1 KO hippocampal neurons with astrocyte-conditioned media of FMRP-expressing (control) astrocytes and the exogenous application of TSP-1, prevented dendritic spine defects in Fmr1 KO neurons. This indicates that astrocyte-secreted TSP-1 is a potent modulator of dendritic morphology and that reduced TSP-1 in FXS astrocytes would contribute to dendritic alterations in FXS neurons.
In agreement with these results, an astrocyte-specific Fmr1 KO mouse model shows similar dendritic and cognitive alterations than Fmr1 KO mice [33]. However, restoring FMRP expression specifically in Fmr1 KO astrocytes was not sufficient to restore dendritic and learning deficits associated with FXS suggesting that astrocyte dysfunction does not completely account for FXS pathophysiology.
2.2. Astrocyte Pathology in Down Syndrome (DS)
DS is the most prevalent cause of intellectual disability of genetic origin. It is due to the presence of a third copy of HSA21, which results in deregulated gene expression leading to altered brain function. DS brain manifestations include changes in the volume and connectivity of certain brain regions such as the cerebral cortex, cerebellum, and hippocampus [81][82], and neuroarchitectural alterations such as spine dysgenesis [14][83], decreased spine density [8][9], and dendritic atrophy [7]. These alterations may deeply perturb information processing in structures related to cognitive functions such as hippocampus and cortex [83] and are assumed to underlie some cognitive impairments in DS, ranging from learning difficulties to spatial memory deficits [84].
In DS, there is an increased number of astrocytes [42][57][85] (Figure 2). In post-mortem brains of individuals with DS, astrocytes are more abundant, bigger, and express more astroglial markers (S100β, GFAP) than age-matched controls [42][85]. Similar observations were reported in Ts65Dn, a partial trisomic mouse model for DS [86]. Nevertheless, there are some discrepancies that suggest that GFAP is reduced in particular brain areas [87][88]. DS foetuses have a higher percentage of cells with astrocytic phenotype in the hippocampus [89], thus indicating a shift from neurogenesis to gliogenesis. This neurogenic-to-gliogenic switch was confirmed in induced pluripotent stem cells (iPSCs) from monozygotic twins discordant for trisomy 21 in which a shift towards the astroglial phenotype was detected in the transcriptional signature of DS-iPSC-derived cells, as shown by the increased expression of GFAP, S100β, and Vimentin [90]. HSA21 genes, such as the Dual specificity tyrosine-phosphorylation-regulated kinase 1A (DYRK1A), may play a role through the activation of the astrogliogenic transcription factor signal transducer and activator of transcription (STAT) that subsequently induces precocious astrogliogenesis by switching the neural progenitor fate towards the astroglial phenotype [91]. These findings suggest that gene expression deregulation in DS, and specifically changes in the DYRK1A-STAT signalling pathway, control the onset of the gliogenic switch and favor the neural progenitor cell fate towards astrogliogenesis.
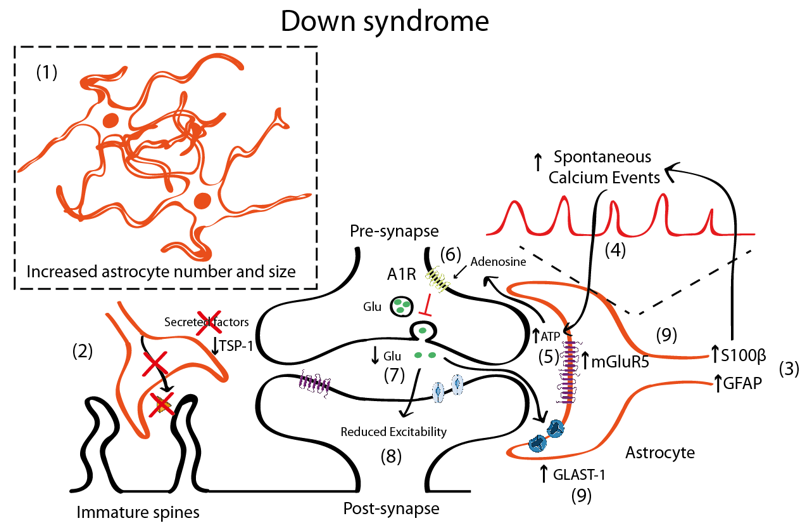
Figure 2. Schematic representation illustrating the astrocyte-synapse alterations in DS. (1) Astrocyte number and volume is increased in DS. (2) Reduced astrocyte secreted TSP-1 prevents spine maturation resulting in more frequent filopodia (immature) spines. (3) Increased S100β and GFAP expression has been described in astrocytes. S100β upregulation has been linked with increased astrocyte calcium oscillations (4). Increased astrocyte activity leads to adenosine triphosphate (ATP) release to the synaptic cleft (5) that is hydrolyzed to adenosine and activates A1 adenosine receptors (A1R) (6). A1R activation prevents glutamate release from the presynaptic terminal and, consequently (7) depresses synaptic transmission (8). Even though mGluR5 is upregulated in astrocytes (and probably in neurons), no mechanistic studies have been performed to uncover the contribution of mGluR5 to DS pathophysiology. (9) Reduced glutamate concentrations can be contributed by increased expression of the glutamate transporter GLAST-1 that leads to increased astroglial glutamate uptake.
The increased astrocyte number [42][57][85] is accompanied by a reduction in the neuronal population [89][92], due to impaired proliferation [89][93] and increased apoptosis [89][94]. This implies that the neuron to glia ratio would be reduced in DS, which could have profound implications. In fact, astrocytes are distributed in non-overlapping synaptic territories in the tridimensional space, which allows to modulate the synaptic transmission of several neurons at a time. These separated anatomical domains, called synaptic islands [95], are particularly relevant, as they might prevent the redundancy of different astrocytes controlling the same or neighbouring synapses. In the DS scenario, astrocytes are increased both in number and in volume while neuronal numbers are reduced. These physical changes may not only affect how neurons and astrocytes are distributed and positioned in the tridimensional space but would also determine how many neurons a single astrocyte contacts. As a consequence, the astrocyte-associated area of influence would be affected, with probable consequences on neuronal communication. We speculate that the changes in the tridimensional arrangement could lead to a synaptic island overlap, promoting a misregulation and redundancy in the astroglial control of synaptic transmission.
In DS, astrocytosis is maintained throughout life and is accompanied by an immature astroglial phenotype with decreased interlaminar processes [96]. Moreover, astroglial physiology is altered, as shown by the increased spontaneous calcium oscillations in DS astrocytes [41]. This may impact neuronal function, since intracellular calcium transients in astrocytes induce the release of gliotransmitters. In fact, DS-iPSCs-derived astrocytes exhibited increased calcium activity, which was shown to subsequently reduce the excitability of co-cultured neurons [41]. The increase of calcium oscillations was attributed to S100β overexpression, since normalisation of S100β expression restored calcium activity to control levels [41]. Interestingly, the reduction of evoked field potentials in DS iPSCs co-cultured neurons was prevented by blocking the A1 adenosine receptor (A1R) with 8-Cyclopentyl-1,3-dipropyl xanthine (DPCPX, a potent A1R antagonist). This would suggest that either A1R is overexpressed in neurons—a fact that to the best of our knowledge has not yet been described. A second possible explanation would be that increased adenosine concentration driven by the hydrolysis of an excess of ATP released consequent to the astrocyte hyperactivity would activate neuronal A1Rs. Adenosine has been shown to activate A1R in neighbouring synapses [97][98] and A1R activation inhibits glutamate release [99][100], consistent with the reduced glutamate levels in DS that lead to an excitatory/inhibitory imbalance [101][102][103]. Specifically, the levels of glutamate are reduced in several brain regions including the parahippocampal gyrus [101] in the hippocampus [102] and in peripheral tissues [103], which could produce an overall reduction in the neuronal activity. These reduced glutamate levels could also be explained by the increased expression of the glutamate transporter GLAST-1 in DS astrocytes leading to a higher glutamate uptake compared to control astroglia [104]. Conversely, the levels of GLT-1 were preserved in DS astrocytes.
Although there is still some controversy, it seems that in DS, there is also an increased inhibition [105][106][107], since blocking GABAA receptors restores LTP deficits in Ts65Dn mice [108], while excitation is preserved [105] or slightly reduced [109]. Some studies suggested that the number of excitatory synapses would be reduced [109], while inhibitory synapses would be preserved [109]. In DS, there is an upregulation of mGluR5 both in fetal and adult DS brains [110] and this mGluR5 upregulation is astrocyte-specific [110]. However, no mechanistic studies associating mGluR5 with synaptic alterations either at the structural or functional level have been performed in mouse models for DS. Nevertheless, the deregulation of mGluR5 signalling in neurons and/or astrocytes can ultimately lead to alterations in the astrocyte–synapse cross-talk that is essential for synaptic transmission and contribute, at least to some extent, to the memory deficits associated with DS.
Interestingly, and similar to the FXS scenario, the co-culture of rat hippocampal neurons that were grown on top of human DS astrocytes showed reduced levels of TSP-1 (around 60% lower compared to WT astrocytes) which led to a reduction in the neuronal spine number and more immature (filopodia) spines compared to neurons cultured with control astrocytes [14][34]. Conversely, the addition of TSP-1 in co-cultures of neurons and DS astrocytes restored the alterations in the dendritic spines, suggesting that TSP-1 dysfunction in DS contributes to aberrant dendritic morphology.
Studies focusing on astrocyte pathology on DS are scarce and limited and very few have explored in detail the mechanisms by which astrocytes could contribute to synaptic alterations in DS. Thus, exploring the mechanisms that might lead to astrocyte–synapse communication such as mGluR5 signalling, purinergic transmission, or deficits in TSP-1 secretion can provide new levels of understanding about the contribution of astrocyte dysfunction to memory deficits in DS.
References
- Dierssen, Top ten discoveries of the year: Neurodevelopmental disorders. Free Neuropathol. 2020, 1, 13. Available online: https://www.uni-muenster.de/Ejournals/index.php/fnp/article/view/2672 (accessed on 26th of October 2020).
- Papazoglou, ; Jacobson, L.A.; McCabe, M.; Kaufmann, W.; Zabel, T.A. To ID or Not to ID? Changes in Classification Rates of Intellectual Disability Using DSM-5. Intellect. Dev. Disabil. 2014, 52, 165–174, doi:10.1352/1934-9556-52.3.165.
- Ardiles, O.; Grabrucker, A.M.; Scholl, F.G.; Rudenko, G.; Borsello, T. Molecular and Cellular Mechanisms of Synaptopathies. Neural Plast. 2017, 2017, 1–3, doi:10.1155/2017/2643943.
- Luo, ; Norris, R.; Gordon, S.; Nithianantharajah, J. Neurodevelopmental synaptopathies: Insights from behaviour in rodent models of synapse gene mutations. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2018, 84, 424–439, doi:10.1016/j.pnpbp.2017.12.001.
- Dierssen, ; Ramakers, G.J.A. Dendritic pathology in mental retardation: From molecular genetics to neurobiology. Genes Brain Behav. 2006, 5, 48–60, doi:10.1111/j.1601-183x.2006.00224.x.
- Irwin, A.; Galvez, R.; Greenough, W.T. Dendritic Spine Structural Anomalies in Fragile-X Mental Retardation Syndrome. Cereb. Cortex 2000, 10, 1038–1044, doi:10.1093/cercor/10.10.1038.
- Dierssen, ; Benavides-Piccione, R.; Martínez-Cué, C.; Estivill, X.; Flórez, J.; Elston, G.; DeFelipe, J. Alterations of neocortical pyramidal cell phenotype in the Ts65Dn mouse model of Down syndrome: Effects of environmental enrichment. Cereb. Cortex 2003, 13, 758–764, doi:10.1093/cercor/13.7.758.
- Belichenko, V.; Masliah, E.; Kleschevnikov, A.M.; Villar, A.J.; Epstein, C.J.; Salehi, A.; Mobley, W.C. Synaptic structural abnormalities in the Ts65Dn mouse model of down syndrome. J. Comp. Neurol. 2004, 480, 281–298, doi:10.1002/cne.20337.
- Catuara-Solarz, ; Espinosa-Carrasco, J.; Erb, I.; Langohr, K.; Gonzalez, J.R.; Notredame, C.; Dierssen, M. Combined Treatment with Environmental Enrichment and (-)-Epigallocatechin-3-Gallate Ameliorates Learning Deficits and Hippocampal Alterations in a Mouse Model of Down Syndrome. eNeuro 2016, 3, doi:10.1523/eneuro.0103-16.2016.
- Comery, A.; Harris, J.B.; Willems, P.J.; Oostra, B.A.; Irwin, S.A.; Weiler, I.J.; Greenough, W.T. Abnormal dendritic spines in fragile X knockout mice: Maturation and pruning deficits. Proc. Natl. Acad. Sci. USA 1997, 94, 5401–5404, doi:10.1073/pnas.94.10.5401.
- Hinton, J.; Brown, W.T.; Wisniewski, K.; Rudelli, R.D. Analysis of neocortex in three males with the fragile X syndrome. Am. J. Med. Genet. 1991, 41, 289–294, doi:10.1002/ajmg.1320410306.
- Xu, ; Miller, E.C.; Pozzo-Miller, L. Dendritic spine dysgenesis in Rett syndrome. Front. Neuroanat. 2014, 8, doi:10.3389/fnana.2014.00097.
- Landi, ; Putignano, E.; Boggio, E.M.; Giustetto, M.; Pizzorusso, T.; Ratto, G.M. The short-time structural plasticity of dendritic spines is altered in a model of Rett syndrome. Sci. Rep. 2011, 1, 45, doi:10.1038/srep00045.
- Garcia, ; Torres, M.; Helguera, P.; Coskun, P.; Busciglio, J. A Role for Thrombospondin-1 Deficits in Astrocyte-Mediated Spine and Synaptic Pathology in Down’s Syndrome. PLoS ONE 2010, 5, e14200, doi:10.1371/journal.pone.0014200.
- Stagni, ; Salvalai, M.E.; Giacomini, A.; Emili, M.; Uguagliati, B.; Xia, E.; Grilli, M.; Bartesaghi, R.; Bartesaghi, R. Neonatal treatment with cyclosporine A restores neurogenesis and spinogenesis in the Ts65Dn model of Down syndrome. Neurobiol. Dis. 2019, 129, 44–55, doi:10.1016/j.nbd.2019.05.005.
- Jacobs, ; Doering, L.C. Astrocytes Prevent Abnormal Neuronal Development in the Fragile X Mouse. J. Neurosci. 2010, 30, 4508–4514, doi:10.1523/jneurosci.5027-09.2010.
- Fukuda, ; Itoh, M.; Ichikawa, T.; Washiyama, K.; Goto, Y.-I. Delayed Maturation of Neuronal Architecture and Synaptogenesis in Cerebral Cortex ofMecp2-Deficient Mice. J. Neuropathol. Exp. Neurol. 2005, 64, 537–544, doi:10.1093/jnen/64.6.537.
- Kleschevnikov, M.; Belichenko, P.V.; Villar, A.J.; Epstein, C.J.; Malenka, R.C.; Mobley, W.C. Hippocampal Long-Term Potentiation Suppressed by Increased Inhibition in the Ts65Dn Mouse, a Genetic Model of Down Syndrome. J. Neurosci. 2004, 24, 8153–8160, doi:10.1523/jneurosci.1766-04.2004.
- Zhao, -G.; Toyoda, H.; Ko, S.W.; Ding, H.-K.; Wu, L.-J.; Zhuo, M. Deficits in Trace Fear Memory and Long-Term Potentiation in a Mouse Model for Fragile X Syndrome. J. Neurosci. 2005, 25, 7385–7392, doi:10.1523/jneurosci.1520-05.2005.
- Martin, G.S.; Lassalle, O.; Brown, J.T.; Manzoni, O.J. Age-Dependent Long-Term Potentiation Deficits in the Prefrontal Cortex of theFmr1Knockout Mouse Model of Fragile X Syndrome. Cereb. Cortex 2015, 26, 2084–2092, doi:10.1093/cercor/bhv031.
- Weng, -M.; McLeod, F.; Bailey, M.E.S.; Cobb, S.R. Synaptic plasticity deficits in an experimental model of rett syndrome: Long-term potentiation saturation and its pharmacological reversal. Neuroscience 2011, 180, 314–321, doi:10.1016/j.neuroscience.2011.01.061.
- Fred Attneave, B.; Hebb, D.O. The Organization of Behavior; A Neuropsychological Theory. Am. J. Psychol. 1950, 63, 633, doi:10.2307/1418888.
- Liu, ; Ramirez, S.; Pang, P.T.; Puryear, C.B.; Govindarajan, A.; Deisseroth, K.; Tonegawa, S. Optogenetic stimulation of a hippocampal engram activates fear memory recall. Nat. Cell Biol. 2012, 484, 381–385, doi:10.1038/nature11028.
- De La Torre, ; De Sola, S.; Pons, M.; Duchon, A.; De Lagran, M.M.; Farré, M.; Fitó, M.; Benejam, B.; Langohr, K.; Rodriguez, J.; et al. Epigallocatechin-3-gallate, a DYRK1A inhibitor, rescues cognitive deficits in Down syndrome mouse models and in humans. Mol. Nutr. Food Res. 2014, 58, 278–288, doi:10.1002/mnfr.201300325.
- De La Torre, ; De Sola, S.; Hernandez, G.; Farré, M.; Pujol, J.; Rodriguez, J.; Espadaler, J.M.; Langohr, K.; Cuenca-Royo, A.; Principe, A.; et al. Safety and efficacy of cognitive training plus epigallocatechin-3-gallate in young adults with Down’s syndrome (TESDAD): A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2016, 15, 801–810, doi:10.1016/s1474-4422(16)30034-5.
- Jacquemont, ; Berry-Kravis, E.; Hagerman, R.; Von Raison, F.; Gasparini, F.; Apostol, G.; Ufer, M.; Portes, V.D.; Gomez-Mancilla, B. The challenges of clinical trials in fragile X syndrome. Psychopharmacology 2014, 231, 1237–1250, doi:10.1007/s00213-013-3289-0.
- Perea, ; Araque, A. Astrocytes Potentiate Transmitter Release at Single Hippocampal Synapses. Science 2007, 317, 1083–1086, doi:10.1126/science.1144640.
- Adamsky, ; Kol, A.; Kreisel, T.; Doron, A.; Ozeri-Engelhard, N.; Melcer, T.; Refaeli, R.; Horn, H.; Regev, L.; Groysman, M.; et al. Astrocytic Activation Generates De Novo Neuronal Potentiation and Memory Enhancement. Cell 2018, 174, 59–71.e14, doi:10.1016/j.cell.2018.05.002.
- Kol, ; Adamsky, A.; Groysman, M.; Kreisel, T.; London, M.; Goshen, I. Astrocytes contribute to remote memory formation by modulating hippocampal–cortical communication during learning. Nat. Neurosci. 2020, 23, 1229–1239, doi:10.1038/s41593-020-0679-6.
- Lee, S.; Ghetti, A.; Pinto-Duarte, A.; Wang, X.; Dziewczapolski, G.; Galimi, F.; Huitron-Resendiz, S.; Piña-Crespo, J.C.; Roberts, A.J.; Verma, I.M.; et al. Astrocytes contribute to gamma oscillations and recognition memory. Proc. Natl. Acad. Sci. USA 2014, 111, E3343–E3352, doi:10.1073/pnas.1410893111.
- Suzuki, ; Stern, S.A.; Bozdagi, O.; Huntley, G.W.; Walker, R.H.; Magistretti, P.J.; Alberini, C.M. Astrocyte-Neuron Lactate Transport Is Required for Long-Term Memory Formation. Cell 2011, 144, 810–823, doi:10.1016/j.cell.2011.02.018.
- Cheng, ; Lau, S.K.M.; Doering, L.C. Astrocyte-secreted thrombospondin-1 modulates synapse and spine defects in the fragile X mouse model. Mol. Brain 2016, 9, 1–15, doi:10.1186/s13041-016-0256-9.
- Hodges, L.; Yu, X.; Gilmore, A.; Bennett, H.; Tjia, M.; Perna, J.F.; Chen, C.-C.; Li, X.; Lu, J.; Zuo, Y. Astrocytic Contributions to Synaptic and Learning Abnormalities in a Mouse Model of Fragile X Syndrome. Biol. Psychiatry 2017, 82, 139–149, doi:10.1016/j.biopsych.2016.08.036.
- Torres, D.; Garcia, O.; Tang, C.; Busciglio, J. Dendritic spine pathology and thrombospondin-1 deficits in Down syndrome. Free. Radic. Biol. Med. 2017, 114, 10–14, doi:10.1016/j.freeradbiomed.2017.09.025.
- Dossi, ; Vasile, F.; Rouach, N. Human astrocytes in the diseased brain. Brain Res. Bull. 2018, 136, 139–156, doi:10.1016/j.brainresbull.2017.02.001.
- Simpson, ; Ince, P.; Lace, G.; Forster, G.; Shaw, P.; Matthews, F.; Savva, G.; Brayne, C.; Wharton, S.B. Astrocyte phenotype in relation to Alzheimer-type pathology in the ageing brain. Neurobiol. Aging 2010, 31, 578–590, doi:10.1016/j.neurobiolaging.2008.05.015.
- Kuchibhotla, V.; Lattarulo, C.R.; Hyman, B.T.; Bacskai, B.J. Synchronous Hyperactivity and Intercellular Calcium Waves in Astrocytes in Alzheimer Mice. Science 2009, 323, 1211–1215, doi:10.1126/science.1169096.
- Santello, ; Toni, N.; Volterra, A. Astrocyte function from information processing to cognition and cognitive impairment. Nat. Neurosci. 2019, 22, 154–166, doi:10.1038/s41593-018-0325-8.
- Blanco-Suárez, ; Caldwell, A.L.M.; Allen, N.J. Role of astrocyte-synapse interactions in CNS disorders. J. Physiol. 2017, 595, 1903–1916, doi:10.1113/jp270988.
- Bally, P.; Farmer, W.T.; Jones, E.V.; Jessa, S.; Kacerovsky, J.B.; Mayran, A.; Peng, H.; Lefebvre, J.L.; Drouin, J.; Hayer, A.; et al. Human iPSC-derived Down syndrome astrocytes display genome-wide perturbations in gene expression, an altered adhesion profile, and increased cellular dynamics. Hum. Mol. Genet. 2020, 29, 785–802, doi:10.1093/hmg/ddaa003.
- Mito, ; Becker, L.E. Developmental Changes of S-100 Protein and Glial Fibrillary Acidic Protein in the Brain in Down Syndrome. Exp. Neurol. 1993, 120, 170–176, doi:10.1006/exnr.1993.1052.
- J∅Rgensen, O.S.; Brooksbank, W.; Balazs, R. Neuronal plasticity and astrocytic reaction in Down syndrome and Alzheimer disease. J. Neurol. Sci. 1990, 98, 63–79, doi:10.1016/0022-510x(90)90182-m.
- Quinlan, A.; Brenner, M.; Goldman, J.E.; Messing, A. GFAP and its role in Alexander disease. Exp. Cell Res. 2007, 313, 2077–2087, doi:10.1016/j.yexcr.2007.04.004.
- Laurence, A.; Fatemi, S.H. Glial fibrillary acidic protein is elevated in superior frontal, parietal and cerebellar cortices of autistic subjects. Cerebellum 2005, 4, 206–210, doi:10.1080/14734220500208846.
- Mizuno, O.; Wang, Y.; Shi, G.; Wang, Y.; Sun, J.; Papadopoulos, S.; Broussard, G.J.; Unger, E.K.; Deng, W.; Weick, J.; et al. Aberrant Calcium Signaling in Astrocytes Inhibits Neuronal Excitability in a Human Down Syndrome Stem Cell Model. Cell Rep. 2018, 24, 355–365, doi:10.1016/j.celrep.2018.06.033.
- Wu, ; Guo, Z.; Gearing, M.; Chen, G. Tonic inhibition in dentate gyrus impairs long-term potentiation and memory in an Alzheimer’s disease model. Nat. Commun. 2014, 5, 1–13, doi:10.1038/ncomms5159.
- Jo, ; Yarishkin, O.; Hwang, Y.J.; Chun, Y.E.; Park, M.; Woo, D.H.; Bae, J.Y.; Kim, T.; Lee, J.; Chun, H.; et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 2014, 20, 886–896, doi:10.1038/nm.3639.
- Gerlai, ; Wojtowicz, J.M.; Marks, A.; Roder, J. Overexpression of a calcium-binding protein, S100 beta, in astrocytes alters synaptic plasticity and impairs spatial learning in transgenic mice. Learn. Mem. 1995, 2, 26–39, doi:10.1101/lm.2.1.26.
- Araujo, B.H.S.; Kaid, C.; De Souza, J.S.; Da Silva, S.G.; Goulart, E.; Caires, L.C.J.; Musso, C.M.; Torres, L.B.; Ferrasa, A.; Herai, R.; et al. Down Syndrome iPSC-Derived Astrocytes Impair Neuronal Synaptogenesis and the mTOR Pathway In Vitro. Mol. Neurobiol. 2017, 55, 5962–5975, doi:10.1007/s12035-017-0818-6.
- Murphy, G.M.; Ellis, W.G.; Lee, Y.-L.; Stultz, K.E.; Shrivastava, R.; Tinklenberg, J.R.; Eng, L.F. Chapter 40: Astrocytic Gliosis in the Amygdala in Down’s Syndrome and Alzheimer’s Disease; Elsevier: Amsterdam, The Netherlands, 1992; Volume 94, pp. 475–483.
- Guttenplan, K.A.; Stafford, B.K.; El-Danaf, R.N.; Adler, D.I.; Münch, A.E.; Weigel, M.K.; Huberman, A.D.; Liddelow, S.A. Neurotoxic Reactive Astrocytes Drive Neuronal Death after Retinal Injury. Cell Rep. 2020, 31, 107776, doi:10.1016/j.celrep.2020.107776.
- Kia, A.; McAvoy, K.; Krishnamurthy, K.; Trotti, D.; Pasinelli, P. Astrocytes expressing ALS-linked mutant FUS induce motor neuron death through release of tumor necrosis factor-alpha. Glia 2018, 66, 1016–1033, doi:10.1002/glia.23298.
- Sofroniew, M. Astrogliosis. Cold Spring Harb. Perspect. Biol. 2015, 7, a020420, doi:10.1101/cshperspect.a020420.
- Pekny, M.; Pekna, M. Astrocyte Reactivity and Reactive Astrogliosis: Costs and Benefits. Physiol. Rev. 2014, 94, 1077–1098, doi:10.1152/physrev.00041.2013.
- Sofroniew, M. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009, 32, 638–647, doi:10.1016/j.tins.2009.08.002.
- Mito, T.; Becker, L.E. Developmental Changes of S-100 Protein and Glial Fibrillary Acidic Protein in the Brain in Down Syndrome. Exp. Neurol. 1993, 120, 170–176, doi:10.1006/exnr.1993.1052.
- Griffin, W.; Sheng, J.G.; McKenzie, J.E.; Royston, M.C.; Gentleman, S.M.; Brumback, R.A.; Cork, L.C.; Del Bigio, M.R.; Roberts, G.W.; Mrak, R.E. Life-long Overexpression of S100β in Down’s Syndrome: Implications for Alzheimer Pathogenesis. Neurobiol. Aging 1999, 19, 401–405, doi:10.1016/s0197-4580(98)00074-8.
- Banerjee, A.; Ifrim, M.F.; Valdez, A.N.; Raj, N.; Bassell, G.J. Aberrant RNA translation in fragile X syndrome: From FMRP mechanisms to emerging therapeutic strategies. Brain Res. 2018, 1693, 24–36, doi:10.1016/j.brainres.2018.04.008.
- Dockendorff, T.C.; Labrador, M. The Fragile X Protein and Genome Function. Mol. Neurobiol. 2018, 56, 711–721, doi:10.1007/s12035-018-1122-9.
- Reiss, A.L.; Aylward, E.; Freund, L.S.; Joshi, P.K.; Bryan, R.N. Neuroanatomy of fragile X syndrome: The posterior fossa. Ann. Neurol. 1991, 29, 26–32, doi:10.1002/ana.410290107.
- Sabaratnam, M. Pathological and neuropathological findings in two males with fragile-X syndrome. J. Intellect. Disabil. Res. 2000, 44, 81–85, doi:10.1046/j.1365-2788.2000.00261.x.
- The Dutch-Belgian Fragile X Consorthrum. Fmr1 knockout mice: A model to study fragile X mental retardation. Cell 1994, 78, doi:10.1016/0092-8674(94)90569-x.
- Higashimori, H.; Morel, L.; Huth, J.; Lindemann, L.; Dulla, C.; Taylor, A.; Freeman, M.; Yang, Y. Astroglial FMRP-dependent translational down-regulation of mGluR5 underlies glutamate transporter GLT1 dysregulation in the fragile X mouse. Hum. Mol. Genet. 2013, 22, 2041–2054, doi:10.1093/hmg/ddt055.
- Bear, M.F.; Huber, K.M.; Warren, S.T. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004, 27, 370–377, doi:10.1016/j.tins.2004.04.009.
- Dölen, G.; Osterweil, E.; Rao, B.S.S.; Smith, G.B.; Auerbach, B.D.; Chattarji, S.; Bear, M.F. Correction of Fragile X Syndrome in Mice. Neuron 2007, 56, 955–962, doi:10.1016/j.neuron.2007.12.001.
- Veloz, M.F.V.; Buijsen, R.A.; Willemsen, R.; Cupido, A.; Bosman, L.W.; Koekkoek, S.K.E.; Potters, J.W.; Oostra, B.A.; De Zeeuw, C.I. The effect of an mGluR5 inhibitor on procedural memory and avoidance discrimination impairments in Fmr1 KO mice. Genes Brain Behav. 2012, 11, 325–331, doi:10.1111/j.1601-183X.2011.00763.x.
- Pop, A.S.; Levenga, J.; De Esch, C.E.F.; Buijsen, R.A.; Nieuwenhuizen, I.M.; Li, T.; Isaacs, A.; Gasparini, F.; Oostra, B.A.; Willemsen, R. (Rob) Rescue of dendritic spine phenotype in Fmr1 KO mice with the mGluR5 antagonist AFQ056/Mavoglurant. Psychopharmacology 2012, 231, 1227–1235, doi:10.1007/s00213-012-2947-y.
- Aloisi, E.; Le Corf, K.; Dupuis, J.; Zhang, P.; Ginger, M.; Labrousse, V.; Spatuzza, M.; Haberl, M.G.; Costa, L.; Shigemoto, R.; et al. Altered surface mGluR5 dynamics provoke synaptic NMDAR dysfunction and cognitive defects in Fmr1 knockout mice. Nat. Commun. 2017, 8, 1103, doi:10.1038/s41467-017-01191-2.
- Carroll, R.C.; Lissin, D.V.; Von Zastrow, M.; Nicoll, R.A.; Malenka, R.C. Rapid redistribution of glutamate receptors contributes to long-term depression in hippocampal cultures. Nat. Neurosci. 1999, 2, 454–460, doi:10.1038/8123.
- Snyder, E.M.; Philpot, B.D.; Huber, K.M.; Dong, X.; Fallon, J.R.; Bear, M.F. Internalization of ionotropic glutamate receptors in response to mGluR activation. Nat. Neurosci. 2001, 4, 1079–1085, doi:10.1038/nn746.
- Zakharenko, S.S.; Zablow, L.; Siegelbaum, S.A. Altered presynaptic vesicle release and cycling during mGluR-dependent LTD. Neuron 2002, 35, 1099–1110, doi:10.1016/s0896-6273(02)00898-x.
- Huber, K.M.; Gallagher, S.M.; Warren, S.T.; Bear, M.F. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. USA 2002, 99, 7746–7750, doi:10.1073/pnas.122205699.
- Yuskaitis, C.J.; Beurel, E.; Jope, R.S. Evidence of reactive astrocytes but not peripheral immune system activation in a mouse model of Fragile X syndrome. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2010, 1802, 1006–1012, doi:10.1016/j.bbadis.2010.06.015.
- Pacey, L.K.K.; Guan, S.; Tharmalingam, S.; Thomsen, C.; Hampson, D.R. Persistent astrocyte activation in the fragile X mouse cerebellum. Brain Behav. 2015, 5, e00400, doi:10.1002/brb3.400.
- Cao, Z.; Hulsizer, S.; Cui, Y.; Pretto, D.L.; Kim, K.H.; Hagerman, P.J.; Tassone, F.; Pessah, I.N. Enhanced Asynchronous Ca2+Oscillations Associated with Impaired Glutamate Transport in Cortical Astrocytes ExpressingFmr1Gene Premutation Expansion. J. Biol. Chem. 2013, 288, 13831–13841, doi:10.1074/jbc.m112.441055.
- Asch, A.S.; Leung, L.L.; Shapiro, J.; Nachman, R.L. Human brain glial cells synthesize thrombospondin. Proc. Natl. Acad. Sci. USA 1986, 83, 2904–2908, doi:10.1073/pnas.83.9.2904.
- Christopherson, K.S.; Ullian, E.M.; Stokes, C.C.; Mullowney, C.E.; Hell, J.W.; Agah, A.; Lawler, J.; Mosher, D.F.; Bornstein, P.; Barres, B.A. Thrombospondins are Astrocyte-Secreted Proteins that Promote CNS Synaptogenesis. Cell 2005, 120, 421–433, doi:10.1016/j.cell.2004.12.020.
- Yu, K.; Ge, J.; Summers, J.B.; Li, F.; Liu, X.; Ma, P.; Kaminski, J.; Zhuang, J. TSP-1 Secreted by Bone Marrow Stromal Cells Contributes to Retinal Ganglion Cell Neurite Outgrowth and Survival. PLoS ONE 2008, 3, e2470, doi:10.1371/journal.pone.0002470.
- Adams, J.C.; Tucker, R.P. The thrombospondin type 1 repeat (TSR) superfamily: Diverse proteins with related roles in neuronal development. Dev. Dyn. 2000, doi:10.1002/(SICI)1097-0177(200006)218:23.0.CO;2-0.
- Lu, Z.; Kipnis, J. Thrombospondin 1—A key astrocyte‐derived neurogenic factor. FASEB J. 2010, 24, 1925–1934, doi:10.1096/fj.09-150573.
- Pinter, J.D.; Eliez, S.; Schmitt, J.E.; Capone, G.T.; Reiss, A.L. Neuroanatomy of Down’s Syndrome: A High-Resolution MRI Study. Am. J. Psychiatry 2001, 158, 1659–1665, doi:10.1176/appi.ajp.158.10.1659.
- Raz, N.; Torres, I.J.; Briggs, S.D.; Spencer, W.D.; Thornton, A.E.; Loken, W.J.; Gunning, F.M.; McQuain, J.D.; Driesen, N.R.; Acker, J.D. Selective neuroanatornic abnormalities in Down’s syndrome and their cognitive correlates: Evidence from MRI morphometry. Neurology 1995, 45, 356–366, doi:10.1212/wnl.45.2.356.
- De Lagran, M.M.; Benavides-Piccione, R.; Ballesteros-Yanez, I.; Calvo, M.; Morales, M.; Fillat, C.; DeFelipe, J.; Ramakers, G.J.A.; Dierssen, M. Dyrk1A Influences Neuronal Morphogenesis Through Regulation of Cytoskeletal Dynamics in Mammalian Cortical Neurons. Cereb. Cortex 2012, 22, 2867–2877, doi:10.1093/cercor/bhr362.
- Lott, I.; Dierssen, M. Cognitive deficits and associated neurological complications in individuals with Down’s syndrome. Lancet Neurol. 2010, 9, 623–633, doi:10.1016/s1474-4422(10)70112-5.
- Griffin, W.S.; Stanley, L.C.; Ling, C.; White, L.; MacLeod, V.; Perrot, L.J.; White, C.L.; Araoz, C. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc. Natl. Acad. Sci. USA 1989, 86, 7611–7615, doi:10.1073/pnas.86.19.7611.
- Lockrow, J.P.; Fortress, A.M.; Granholm, A.-C.E. Age-Related Neurodegeneration and Memory Loss in Down Syndrome. Curr. Gerontol. Geriatr. Res. 2012, 2012, 1–13, doi:10.1155/2012/463909.
- Goodison, K.L.; Parhad, I.M.; White, C.L.; Sima, A.A.F.; Clark, A.W. Neuronal and Glial Gene Expression in Neocortex of Downʼs Syndrome and Alzheimerʼs Disease. J. Neuropathol. Exp. Neurol. 1993, 52, 192–198, doi:10.1097/00005072-199305000-00002.
- Kanaumi, T.; Milenkovic, I.; Adle‐Biassette, H.; Aronica, E.; Kovacs, G.G. Non‐neuronal cell responses differ between normal and Down syndrome developing brains. Int. J. Dev. Neurosci. 2013, 31, 796–803, doi:10.1016/j.ijdevneu.2013.09.011.
- Guidi, S.; Bonasoni, P.; Ceccarelli, C.; Santini, D.; Gualtieri, F.; Ciani, E.; Bartesaghi, R. RESEARCH ARTICLE: Neurogenesis Impairment and Increased Cell Death Reduce Total Neuron Number in the Hippocampal Region of Fetuses with Down Syndrome. Brain Pathol. 2007, 18, 180–197, doi:10.1111/j.1750-3639.2007.00113.x.
- Hibaoui, Y.; Grad, I.; Letourneau, A.; Sailani, M.R.; Dahoun, S.; Santoni, F.A.; Gimelli, S.; Guipponi, M.; Pelte, M.F.; Bena, F.S.; et al. Modelling and rescuing neurodevelopmental defect of D own syndrome using induced pluripotent stem cells from monozygotic twins discordant for trisomy 21. EMBO Mol. Med. 2013, 6, 259–277, doi:10.1002/emmm.201302848.
- Kurabayashi, N.; Nguyen, M.D.; Sanada, K. DYRK 1A overexpression enhances STAT activity and astrogliogenesis in a Down syndrome mouse model. EMBO Rep. 2015, 16, 1548–1562, doi:10.15252/embr.201540374.
- Lorenzi, H.A.; Reeves, R.H. Hippocampal hypocellularity in the Ts65Dn mouse originates early in development. Brain Res. 2006, 1104, 153–159, doi:10.1016/j.brainres.2006.05.022.
- Contestabile, A.; Fíla, T.; Cappellini, A.; Bartesaghi, R.; Ciani, E. Widespread impairment of cell proliferation in the neonate Ts65Dn mouse, a model for Down syndrome. Cell Prolif. 2009, 42, 171–181, doi:10.1111/j.1365-2184.2009.00587.x.
- Anderson, A.J.; Stoltzner, S.; Lai, F.; Su, J.; Nixon, R.A. Morphological and biochemical assessment of DNA damage and apoptosis in Down syndrome and Alzheimer disease, and effect of postmortem tissue archival on TUNEL. Neurobiol. Aging 2000, 21, 511–524, doi:10.1016/s0197-4580(00)00126-3.
- Halassa, M.M.; Fellin, T.; Takano, H.; Dong, J.-H.; Haydon, P.G. Synaptic Islands Defined by the Territory of a Single Astrocyte. J. Neurosci. 2007, 27, 6473–6477, doi:10.1523/jneurosci.1419-07.2007.
- Colombo, J.A.; Reisin, H.D.; Jones, M.; Bentham, C. Development of interlaminar astroglial processes in the cerebral cortex of control and Down’s syndrome human cases. Exp. Neurol. 2005, 193, 207–217, doi:10.1016/j.expneurol.2004.11.024.
- Zhang, J.-M.; Wang, H.-K.; Ye, C.-Q.; Ge, W.; Chen, Y.; Jiang, Z.-L.; Wu, C.-P.; Poo, M.-M.; Duan, S. ATP Released by Astrocytes Mediates Glutamatergic Activity-Dependent Heterosynaptic Suppression. Neuron 2003, 40, 971–982, doi:10.1016/s0896-6273(03)00717-7.
- Pascual, O.; Casper, K.B.; Kubera, C.; Zhang, J.; Revilla-Sanchez, R.; Sul, J.-Y.; Takano, H.; Moss, S.J.; McCarthy, K.; Haydon, P.G. Astrocytic Purinergic Signaling Coordinates Synaptic Networks. Science 2005, 310, 113–116, doi:10.1126/science.1116916.
- Dunwiddie, T.V.; Masino, S.A. The Role and Regulation of Adenosine in the Central Nervous System. Annu. Rev. Neurosci. 2001, 24, 31–55, doi:10.1146/annurev.neuro.24.1.31.
- Lindquist, B.E.; Shuttleworth, C.W. Adenosine receptor activation is responsible for prolonged depression of synaptic transmission after spreading depolarization in brain slices. Neuroscience 2012, 223, 365–376, doi:10.1016/j.neuroscience.2012.07.053.
- Risser, D.; Lubec, G.; Cairns, N.; Herrera-Marschitz, M. Excitatory amino acids and monoamines in parahippocampal gyrus and frontal cortical pole of adults with down syndrome. Life Sci. 1997, 60, 1231–1237, doi:10.1016/s0024-3205(97)00067-2.
- Reynolds, G.P.; Warner, C.E. Amino acid neurotransmitter deficits in adult Down’s syndrome brain tissue. Neurosci. Lett. 1988, 94, 224–227, doi:10.1016/0304-3940(88)90299-6.
- Begni, B.; Brighina, L.; Fumagalli, L.; Andreoni, S.; Castelli, E.; Francesconi, C.; Del Bo, R.; Bresolin, N.; Ferrarese, C. Altered glutamate uptake in peripheral tissues from Down Syndrome patients. Neurosci. Lett. 2003, 343, 73–76, doi:10.1016/s0304-3940(03)00260-x.
- Chen, C.; Jiang, P.; Xue, H.; Peterson, S.E.; Tran, H.T.; McCann, A.E.; Parast, M.M.; Li, S.; Pleasure, D.E.; Laurent, L.C.; et al. Role of astroglia in Down’s syndrome revealed by patient-derived human-induced pluripotent stem cells. Nat. Commun. 2014, 5, 4430, doi:10.1038/ncomms5430.
- Belichenko, P.V.; Kleschevnikov, A.M.; Masliah, E.; Wu, C.; Takimoto-Kimura, R.; Salehi, A.; Mobley, W.C. Excitatory-inhibitory relationship in the fascia dentata in the Ts65Dn mouse model of down syndrome. J. Comp. Neurol. 2009, 512, 453–466, doi:10.1002/cne.21895.
- Harashima, C.; Jacobowitz, D.M.; Stoffel, M.; Chakrabarti, L.; Haydar, T.F.; Siarey, R.J.; Galdzicki, Z. Elevated Expression of the G-Protein-Activated Inwardly Rectifying Potassium Channel 2 (GIRK2) in Cerebellar Unipolar Brush Cells of a Down Syndrome Mouse Model. Cell. Mol. Neurobiol. 2006, 26, 717–732, doi:10.1007/s10571-006-9066-4.
- Best, T.K.; Cramer, N.P.; Chakrabarti, L.; Haydar, T.F.; Galdzicki, Z. Dysfunctional hippocampal inhibition in the Ts65Dn mouse model of Down syndrome. Exp. Neurol. 2011, 233, 749–757, doi:10.1016/j.expneurol.2011.11.033.
- Costa, A.C.; Grybko, M.J. Deficits in hippocampal CA1 LTP induced by TBS but not HFS in the Ts65Dn mouse: A model of Down syndrome. Neurosci. Lett. 2005, 382, 317–322, doi:10.1016/j.neulet.2005.03.031.
- Kurt, M.; Davies, D.C.; Kidd, M.; Dierssen, M.; Flórez, J. Synaptic deficit in the temporal cortex of partial trisomy 16 (Ts65Dn) mice. Brain Res. 2000, 858, 191–197, doi:10.1016/s0006-8993(00)01984-3.
- Oka, A.; Takashima, S. The up-regulation of metabotropic glutamate receptor 5 (mGluR5) in Down’s syndrome brains. Acta Neuropathol. 1999, 97, 275–278, doi:10.1007/s004010050985.

