 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Camila Xu | -- | 1477 | 2022-11-15 01:35:22 |
Video Upload Options
Polony sequencing is an inexpensive but highly accurate multiplex sequencing technique that can be used to “read” millions of immobilized DNA sequences in parallel. This technique was first developed by Dr. George Church's group at Harvard Medical School. Unlike other sequencing techniques, Polony sequencing technology is an open platform with freely downloadable, open source software and protocols. Also, the hardware of this technique can be easily set up with a commonly available epifluorescence microscopy and a computer-controlled flowcell/fluidics system. Polony sequencing is generally performed on paired-end tags library that each molecule of DNA template is of 135 bp in length with two 17–18 bp paired genomic tags separated and flanked by common sequences. The current read length of this technique is 26 bases per amplicon and 13 bases per tag, leaving a gap of 4–5 bases in each tag.
1. Workflow
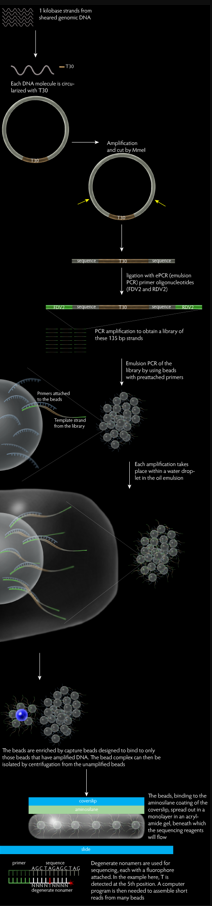
The protocol of Polony sequencing can be broken into three main parts, which are the paired end-tag library construction, template amplification and DNA sequencing.
1.1. Paired End-Tag Library Construction
This protocol begins by randomly shearing the tested genomic DNA into a tight size distribution. The sheared DNA molecules are then subjected for the end repair and A-tailed treatment. The end repair treatment converts any damaged or incompatible protruding ends of DNA to 5’-phosphorylated and blunt-ended DNA, enabling immediate blunt-end ligation, while the A-tailing treatment adds an A to the 3’ end of the sheared DNA. DNA molecules with a length of 1 kb are selected by loading on the 6% TBE PAGE gel. In the next step, the DNA molecules are circularized with T-tailed 30 bp long synthetic oligonucleotides (T30), which contains two outward-facing MmeI recognition sites, and the resulting circularized DNA undergoes rolling circle replication. The amplified circularized DNA molecules are then digested with MmeI (type IIs restriction endonuclease) which will cuts at a distance from its recognition site, releasing the T30 fragment flanked by 17–18 bp tags (≈70 bp in length). The paired-tag molecules need to be end-repaired prior to the ligation of ePCR (emulsion PCR) primer oligonucleotides (FDV2 and RDV2) to their both ends. The resulting 135 bp library molecules are size-selected and nick translated. Lastly, amplify the 135 bp paired end-tag library molecules with PCR to increase the amount of library material and eliminate extraneous ligation products in a single step. The resulted DNA template consists of a 44 bp FDV sequence, a 17–18 bp proximal tag, the T30 sequence, a 17-18 bp distal tag, and a 25 bp RDV sequence.
1.2. Template Amplification
Emulsion PCR
The mono-sized, paramagnetic streptavidin–coated beads are pre-loaded with dual biotin forward primer. Streptavidin has a very strong affinity for biotin, thus the forward primer will bind firmly on the surface of the beads. Next, an aqueous phase is prepared with the pre-loaded beads, PCR mixture, forward and reverse primers, and the paired end-tag library. This is mixed and vortexed with an oil phase to create the emulsion. Ideally, each droplet of water in the oil emulsion has one bead and one molecule of template DNA, permitting millions of non-interacting amplification within a milliliter-scale volume by performing PCR.
Emulsion breaking
After amplification, the emulsion from preceding step is broken using isopropanol and detergent buffer (10 mM Tris pH 7.5, 1 mM EDTA pH 8.0, 100 mM NaCl, 1% (v/v) Triton X‐100, 1% (w/v) SDS), following with a series of vortexing, centrifuging, and magnetic separation. The resulted solution is a suspension of empty, clonal and non-clonal beads, which arise from emulsion droplets that initially have zero, one or multiple DNA template molecules, respectively. The amplified bead could be enriched in the following step.
Bead enrichment
The enrichment of amplified beads is achieved through hybridization to a larger, low density, non-magnetic polystyrene beads that pre-loaded with a biotinylated capture oligonucleotides (DNA sequence that complementary to ePCR amplicon sequence). The mixture is then centrifuged to separate the amplified and capture beads complex from the unamplified beads. The amplified, capture bead complex has a lower density and thus will remain in the supernatant while the unamplified beads form a pellet. The supernatant is recovered and treated with NaOH which will break the complex. The paramagnetic amplified beads are separated from the non-magnetic capture beads by magnetic separation. This enrichment protocol is capable in enriching five times of amplified beads.
Bead capping
The purpose of bead capping is to attach a “capping” oligonucleotide to the 3’ end of both unextended forward ePCR primers and the RDV segment of template DNA. The cap that being use is an amino group that prevents fluorescent probes from ligating to these ends and at the same time, helping the subsequent coupling of template DNA to the aminosilanated flow cell coverslip.
Coverslip arraying
First, the coverslips are washed and aminosilane-treated, enabling the subsequent covalent coupling of template DNA on it and eliminating any fluorescent contamination. The amplified, enriched beads are mixed with acrylamide and poured into a shallow mold formed by a Teflon-masked microscope slide. Immediately, place the aminosilane-treated coverslip on top of the acrylamide gel and allow to polymerize for 45 minutes. Next, invert the slide/coverslip stack and remove the microscope slide from gel. The silane-treated coverslips will bind covalently to the gel while the Teflon on the surface of microscope slide will enable the better removal of slide from the acrylamide gel. The coverslips then bonded to the flow cell body and any unattached beads will be removed.
1.3. DNA Sequencing
The biochemistry of Polony sequencing mainly relies on the discriminatory capacities of ligases and polymerases. First, a series of anchor primers are flowed through the cells and hybridize to the synthetic oligonucleotide sequences at the immediate 3’ or 5’ end of the 17-18 bp proximal or distal genomic DNA tags. Next, an enzymatic ligation reaction of the anchor primer to a population of degenenerate nonamers that are labeled with fluorescent dyes is performed.
Differentially labeled nonamers:
5' Cy5‐NNNNNNNNT
5' Cy3‐NNNNNNNNA
5' TexasRed‐NNNNNNNNC
5' 6FAM‐NNNNNNNNG
The fluorophore-tagged nonamers anneal with differential success to the tag sequences according to a strategy similar to that of degenerate primers, but instead of submission to polymerases, nonamers are selectively ligated onto adjoining DNA- the anchor primer. The fixation of the fluorophore molecule provides a fluorescent signal that indicates whether there is an A, C, G, or T at the query position on the genomic DNA tag. After four-colour imaging, the anchor primer/nonamer complexes are stripped off and a new cycle is begun by replacing the anchor primer. A new mixture of the fluorescently tagged nonamers is introduced, for which the query position is shifted one base further into the genomic DNA tag.
5' Cy5‐NNNNNNNTN
5' Cy3‐NNNNNNNAN
5' TexasRed‐NNNNNNNCN
5' 6FAM‐NNNNNNNGN
Seven bases from the 5’ to 3’ direction and six bases from the 3’ end could be queried in this fashion. The ultimate result is a read length of 26 bases per run (13 bases from each of the paired tags) with a 4-base to 5-base gap in the middle of each tag.
2. Analysis and Software
The polony sequencing generates millions of 26 reads per run and this information needed to be normalized and converted to sequence. These can be done by the software that has been developed by Church Lab. All of the software is free and could be downloaded from the website.[1]
3. Instruments
The sequencing instrument used in this technique could be set up by the commonly available fluorescence microscope and a computer controlled flowcell. The required instruments cost around US$130,000 in 2005. A dedicated polony sequencing machine, Polonator, was developed in 2009 and sold at US$170,000 by Dover.[2][3] It and featured open source software, reagents, and protocols and was intended for use in the Personal Genome Project.[4]
4. Strength and Weaknesses
Polony sequencing allows for a high throughput and high consensus accuracies of DNA sequencing based on a commonly available, inexpensive instrument. Also, it is a very flexible technique that enables variable application including BAC (bacterial artificial chromosome) and bacterial genome resequencing, as well as SAGE (serial analysis of gene expression) tag and barcode sequencing. Furthermore, the polony sequencing technique is emphasized as an open system that shares everything including the software that have been developed, protocol and reagents.
However, although the raw data acquisition could be achieved as high as 786 gigabits but only 1 bit of information out of 10,000 bits collected is useful. Another challenge of this technique is the uniformity of the relative amplification of individual targets. The non-uniform amplification could lower the efficiency of sequencing and posted as the biggest obstacle in this technique.
5. History
Polony sequencing is a development of the polony technology from the late 1990s and 2000s.[5] Methods were developed in 2003 to sequence in situ polonies using single-base extension which could achieve 5-6 bases reads.[6] By 2005, these early attempts had been overhauled to develop the existing polony sequencing technology.[7] The highly parallel sequencing-by-ligation concepts of polony sequencing has contributed the basis for later sequencing technologies such as ABI Solid Sequencing.
References
- "Open Source Sequencing" (in en). http://arep.med.harvard.edu/Polonator/.
- "After Early-Access Phase, Updated Polonator Is Ready for Rollout with $170K Price Tag" (in en). GenomeWeb. 2009-05-05. https://www.genomeweb.com/sequencing/after-early-access-phase-updated-polonator-ready-rollout-170k-price-tag.
- "Dover's Polonator Unaffected by Danaher's Restructuring, Official Says" (in en). GenomeWeb. 2009-09-08. https://www.genomeweb.com/sequencing/dovers-polonator-unaffected-danahers-restructuring-official-says.
- "The Polonator". 2015-04-03. http://www.polonator.org.
- Adessi, C.; Matton, G.; Ayala, G.; Turcatti, G.; Mermod, J. J.; Mayer, P.; Kawashima, E. (2000-10-15). "Solid phase DNA amplification: characterisation of primer attachment and amplification mechanisms". Nucleic Acids Research 28 (20): E87. doi:10.1093/nar/28.20.e87. ISSN 1362-4962. PMID 11024189. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=110803
- Shendure, Jay; Porreca, Gregory J.; Reppas, Nikos B.; Lin, Xiaoxia; McCutcheon, John P.; Rosenbaum, Abraham M.; Wang, Michael D.; Zhang, Kun et al. (2005-09-09). "Accurate Multiplex Polony Sequencing of an Evolved Bacterial Genome" (in en). Science 309 (5741): 1728–1732. doi:10.1126/science.1117389. ISSN 0036-8075. PMID 16081699. https://dx.doi.org/10.1126%2Fscience.1117389
- Shendure, Jay; Porreca, Gregory J.; Reppas, Nikos B.; Lin, Xiaoxia; McCutcheon, John P.; Rosenbaum, Abraham M.; Wang, Michael D.; Zhang, Kun et al. (2005-09-09). "Accurate Multiplex Polony Sequencing of an Evolved Bacterial Genome" (in en). Science 309 (5741): 1728–1732. doi:10.1126/science.1117389. ISSN 0036-8075. PMID 16081699. https://dx.doi.org/10.1126%2Fscience.1117389

