 +1 credit
+1 credit
Video Upload Options
Targeting altered tumour metabolism is an emerging therapeutic strategy for cancer treatment. The metabolic reprogramming that accompanies the development of malignancy creates targetable differences between cancer cells and normal cells, which may be exploited for therapy. In this entry, we focus on the metabolic dysregulation exerted by tumour cells on the immune microenvironment, leading to tumour immunosuppression. This metabolic rewiring and crosstalk with the tumour microenvironment also play a key role in cell proliferation, metastasis, and the development of treatment resistance. Nonetheless, greater understanding of the metabolic crosstalk presents strategies that aid in the precision targeting of altered tumour metabolism, including therapeutic strategies combining metabolic inhibition with immunotherapy.
1. Introduction
To sustain the rapid proliferation characterising cancer cells, corresponding alterations to tumour metabolism must occur to fuel the elevated bioenergetic demands. This understanding has led to the introduction of ‘Deregulating Cellular Energetics’ as a new hallmark of cancer [1]. Initial observations by Otto Warburg described an unusual reliance of cancer cells on glycolysis despite sufficient oxygen, which was later termed the ‘Warburg effect’ to describe this form of aerobic glycolysis [2]. This metabolic reprogramming, while less efficient in terms of ATP production, confers cancer cells with much-needed metabolic intermediates that can be channelled into biosynthetic pathways, such as the pentose-phosphate pathway (PPP) for nucleotide synthesis [3].
While this aerobic ‘Warburg’ glycolytic phenotype had been regarded as the norm in cancer cells, it is becoming increasingly clear that the metabolic needs of tumour cells do not rely on a single metabolic strategy. Recent studies suggest that certain subtypes of cancer cells may preferentially utilize oxidative phosphorylation (OXPHOS) for energy production in glucose-limiting conditions [4]. In addition, OXPHOS dependency may be induced by certain therapies, such as prolonged tyrosine kinase inhibitor (TKI) therapy in certain oncogene-addicted cancers [5][6]. This reflects a phenomenon termed ‘metabolic flexibility’ where cancer cells adjust their metabolic phenotypes in order to gain a selective advantage for cell growth and survival under hostile conditions throughout tumorigenesis up to the time of metastasis [7]. The significance of these metabolic alterations may diverge not only according to intrinsic signalling pathways within the cancer cell, but also rely on the interaction of cancer cells with their surrounding tumour microenvironment (TME), ranging from immune cells and stromal cells to extracellular matrix (ECM) components and soluble factors [8]. There is also emerging evidence to suggest that metabolic reprogramming within cancer stem cell (CSC)-like phenotypes contributes to treatment resistance, therapeutic failure, and cancer relapse.
2. Tumour Immune Microenvironment
The immune system interacts intimately with tumour development in a complex, bidirectional crosstalk that can both inhibit and enhance tumour growth and progression. This interaction has gained recognition as a hallmark of cancer and immunotherapy has become an established pillar of cancer therapy [1]. Immune cells execute their function most effectively when they are able to respond swiftly to environmental stimuli through phenotypic shifts, enhanced by the radical reprogramming of immune cell metabolism [9]. On the other hand, impaired metabolic flexibility results in an ineffective anti-tumour immune response, and may be explained by the mutual metabolic requirements of immune cells and tumour cells, which compete for similar essential nutrients such as glucose and glutamine. Besides nutrient availability, high production of metabolites such as lactate, kynurenine, and other metabolic by-products of cancer metabolism can result in tumour immunosuppression [10].
2.1. T Cells
The effects of altered tumour metabolism on T cell function is summarised in Figure .
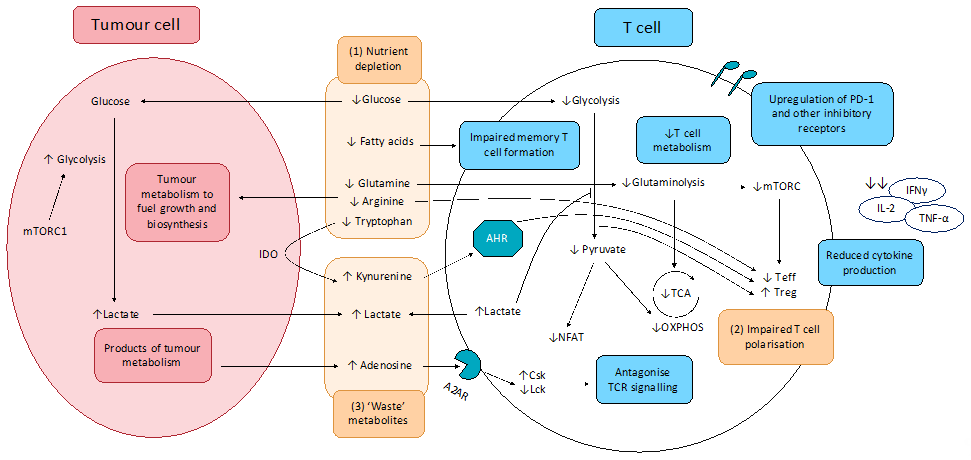
Figure 1. Effect of tumour metabolism on T cell function. (1) Altered cancer cell metabolism results in nutrient competition, depriving T cells of essential nutrients essential for robust anti-tumour activity, including glucose and key amino acids. Resultant exhausted T cell phenotype shows upregulation of inhibitory receptors including PD-1, CTLA-4, TIM-3, LAG-3, and TIGIT, impaired production and release of effector cytokines (IFNγ, IL-2, and TNF-α), as well as impaired degranulation. (2) Depletion of key nutrients and aberrant metabolite signalling promotes pro-tumourigenic T cell phenotypes. (3) Cancer cell metabolism produces lactate and other ‘waste’ metabolites that inhibit T cell function and promote T cell exhaustion.
A2AR: Adenosine 2A receptor; AHR: aryl hydrocarbon receptor; Csk: C-terminal Src kinase; IDO: indoleamine-pyrrole 2,3-dioxygenase; Lck: lymphocyte-specific protein tyrosine kinase; mTORC1: mammalian target of rapamycin complex 1; NFAT: nuclear factor of activated T-cells; OXPHOS: oxidative phosphorylation; TCA: tricarboxylic acid cycle; Teff; effector T cells; Treg: regulatory T cells.
2.1.1. Altered CCM Deprives T cells of Nutrients Essential for Anti-Tumour Activity and Induces Polarisation of Immunosuppressive T Cell Subsets
When a T cell is activated, there is a dramatic metabolic reprogramming mediated through the PI3K/Akt/mTOR pathway, greatly reminiscent of the metabolic reprogramming observed in cancer cells. This ‘Warburg phenotype’ adopted by activated T cells involves upregulation of aerobic glycolysis, increased glucose metabolism through the PPP, increased glutaminolysis, and increased FA synthesis. This leads to a competition between effector T cells (Teff) and tumour cells for similar nutrients especially glucose, thereby impairing the Teff anti-tumour response. Furthermore, glucose limitation is reported to produce an ‘exhausted’ T cell phenotype, characterised by increased programmed cell death protein 1 (PD-1) expression on the surfaces of T cells, which accounts for the greater proportion of ‘exhausted’ T cells in tumours and leads to cancer immune evasion [11][12][13]. Tumour-mediated T cell exhaustion is also characterised by the upregulation of other inhibitory receptors including cytotoxic T lymphocyte-associated antigen 4 (CTLA-4), T-cell immunoglobulin, and mucin-domain containing-3 (TIM-3), lymphocyte activation gene-3 (LAG-3), and T cell immunoreceptor with Ig and immunoreceptor tyrosine-based inhibitory motif (ITIM) domains (TIGIT) [14][15][16][17]. Decreased glucose metabolism was also found to impair the epigenetic reprogramming required for T cell activation. Reduced flux through the glycolytic pathway leads to insufficient acetyl-CoA to maintain α-KG levels required for cofactor function for histone acetylation, resulting in reduced interferon γ (IFNγ) expression and impaired helper T cell (Th1) activity [18].
Similar to glucose, activated T cells have a higher requirement for amino acids, most notably glutamine. Glutamine is utilised by active T cells and is required for the inflammatory responses of Th1 and Th17 cells [19][20], and decreased glutamine availability in the TME is reported to blunt anti-tumour immunity by limiting essential biosynthetic pathways for T cell proliferation. A concern with targeting metabolic pathways is the extensive overlap between the metabolic phenotypes of tumour cells and activated immune cells. Theoretically, GLS inhibition can limit T cell metabolism along with crippling tumour metabolism since increased glutaminolysis is a hallmark of both tumour cells and activated T cells. However, Leone et al., 2019, showed that, while glutamine blockade in cancer cells led to suppression of oxidative and glycolytic metabolism, by contrast CD8+ T cells responded by upregulating acetate metabolism, generating high levels of acetyl-CoA for direct fuelling of the TCA cycle as well as indirect fuelling via increased glucose anaplerosis through pyruvate carboxylase activation. These resulted in upregulation of oxidative metabolism with CD8+ T cells adopting a long-lived, highly activated phenotype [21]. These divergent responses to GLS inhibition serves as a ‘metabolic checkpoint’ and an opportunity to simultaneously inhibit tumour metabolism while boosting anti-tumour immune activity. Other amino acids required for T cell activity includes arginine and tryptophan. Tumour-depletion of arginine in the TME can impair T cell anti-tumour immunity, particularly memory T cell immunity [22]. Tryptophan deficiency is also known to inhibit mTORC1 activity in T cells, impairing T cell activation and proliferation [23].
The effect of FAs is less well-characterised because different T cell subsets utilise FAs differently. Of note, memory T cells are more dependent on FA oxidation (FAO) for energy and are unable to develop in the absence of FAs in culture. However, recent studies suggest that FAs used by memory T cells for FAO are derived from extracellular glucose, rather than direct utilisation of extracellular FAs [24]. Currently, the role of FAs in T cell metabolism is unclear, and further studies are required.
In addition, nutrient depletion in the TME alters T cell differentiation and induces the polarisation of immunosuppressive T cell subsets [25][26]. Glucose deficiency enriches for regulatory T cells (Tregs) because, in contrast to Teffs that rely on aerobic glycolysis, Tregs rely more on FAO. FOXP3 metabolic reprogramming leads to MYC and glycolysis suppression, which enhances OXPHOS and NADH oxidation. These adaptions confer Tregs a metabolic advantage in the low-glucose, high-lactate microenvironment in the TME, shifting the balance to favour Treg enrichment over Teffs and facilitating tumour immune evasion [27]. Glutamine deficiency in the TME may also shift T cell differentiation toward a pro-tumourigenic phenotype, as glutamine deficiency disproportionately impairs Th1 and Th17 subsets more than Tregs, thereby enriching for Tregs in the TME [25].
2.1.2. CCM-Derived ‘Waste’ Metabolites Inhibit T Cell Function and Promotes T Cell Exhaustion
Lactate is reported to inhibit T cell proliferation and cytokine production [28][29]. Tumour-derived lactate accumulates in the TME, leading to impaired T cell export of lactate and intracellular build-up. Elevated lactate suppresses glycolytic enzymes via end-product inhibition, impairing T cell metabolism and function. Lactate build-up in the TME also causes T cell acidification, preventing translocation of nuclear factor of activated T cells (NFAT) into the nucleus and NFAT-mediated transcription. This, thus, inhibits IFNγ production and impairs T cell response [29]. Finally, lactate is reported to inhibit the PI3K/Akt/mTOR pathway in T cells, blunting T cell activation [30][31].
Other tumour-derived ‘waste’ metabolites are also suggested to play a key role in T cell immunosuppression (Figure 1). Adenosine is released by tumours into the TME and inhibits the T cell anti-tumour response. Upon binding to Adenosine A2A receptor (A2AR), signalling leads to an increase in cAMP levels, protein kinase A (PKA) phosphorylation of Csk, which subsequently inhibits Lck and antagonising TCR signalling. This leads to reduced T cell activation, cytokine production, and anti-tumour immunity [32]. Kynurenine, the first breakdown product in indoleamine 2,3-dioxygenase (IDO)-dependent tryptophan degradation, has also been reported to exert immunosuppressive effects and induce T cell apoptosis [23][33].
2.2. Myeloid-Derived Suppressor Cells, Tumour-Associated Macrophages, and Dendritic Cells
Myeloid-derived suppressor cells (MDSCs) are a heterogeneous population of cells of myeloid origin that contribute to TME immunosuppression and exert suppression on T cell and innate immune cell responses. The altered CCM environment influences MDSC functionality, which can further bolster their immunosuppressive effects.
For example, the hypoxic TME leads to HIF-1α signalling, which aids in MDSC differentiation to tumour-promoting tumour-associated macrophages (TAMs) [34]. Lactate similarly induces polarisation toward the pro-tumourigenic M2 macrophage phenotype via HIF-1α signalling [35], and induces upregulation of PD-L1 on myeloid cells, facilitating Teff suppression [36]. Furthermore, hypoxia and lactate in the TME induce a metabolic switch from glycolysis toward OXPHOS, which is consistent with the enrichment and continued functionality of MDSCs and TAMs in a primarily hypoglycaemic TME [37].
The anti-tumour functions of macrophages are also inhibited by altered cancer metabolism. Extracellular lactate reduces activation of monocytes, as measured by reduced glycolysis-dependent tumour necrosis factor (TNF) production [38]. Tumour prostaglandin E2 (PGE2) production is also found to subvert myeloid cell function. This was reported in various oncogene-addicted tumour models [39]. For example, in a BRAF-mutant model of melanoma, and NRAS-mutant models of melanoma, breast, and CRC, PGE2 production impairs myeloid cell activation especially the antigen-presenting ability required for T cell activation [39].
Dendritic cells (DCs) are also key players in anti-tumour immunity. Activation of DCs involves metabolic reprogramming not unlike that of T cells, switching from OXPHOS to aerobic glycolysis. Competition with tumour cells in the TME for essential nutrients, in particular glucose, can severely limit DC activity and antigen-presenting ability [40]. In addition, a low energy state leads to elevated AMPK signalling, which inhibits glycolysis and promotes greater OXPHOS and FAO. This is reminiscent of a tolerogenic DC phenotype [41]. In addition, FAO induction in tumour-associated DCs (TADCs) was found to drive the production of IDO, which results in Treg polarisation and further immunosuppression of T cells in a model of melanoma [42]. Expression of the inhibitory receptor CTLA-4 on Tregs can also induce IDO activity by DCs [43]. This immunosuppressive crosstalk between dysregulated immune cells serve to drive a positive-feedback loop whereby immunosuppression is self-maintained and further propagated in the TME [40].
2.3. Natural Killer Cells and Neutrophils
Other cells of the innate immune system are also intricately linked to metabolic changes in the TME. Due to the greater energetic demands of natural killer (NK) cells, in particular increased glycolysis, NK cells are also subject to competition with tumour cells for glucose. Thus, the perennial problem of glucose and nutrient deprivation in the TME also impairs NK function. Furthermore, the aberrant production of metabolites as a consequence of altered CCM also impacts NK activity. Metabolic reprogramming of NK cells upon activation requires the SREBP transcription factors. 25-hydroxycholesterol (25-HC) is a cholesterol-derived metabolite produced by various cancers, such as glioblastoma [44], and can inhibit translocation of SREBP from the ER to the Golgi, impairing NK activation [45]. Elevated lactate in the TME also reduces NFAT signalling in NK cells, reducing IFNγ production, CD25 levels, and tumour-killing capabilities [29].
Neutrophils are frequently discounted from a metabolic perspective as purely glycolytic. The low glucose availability in the TME is predicted to limit neutrophil ROS production, which can disrupt CD4+ T cell viability and function. However, tumour-directed metabolic reprogramming can switch neutrophils to an oxidative phenotype. For instance, 4T1 tumours was found to induce a metabolic shift to produce mitochondria-rich oxidative neutrophils through aberrant stem cell factor (SCF)/c-Kit signalling. Oxidative neutrophils can use mitochondrial FAO to support NADPH oxidase-dependent ROS production in the hypoglycaemic TME. Thus, tumour-mediated SCF/c-Kit signalling can induce an oxidative phenotype in neutrophils to overcome to metabolic limitations, resulting in maintained ROS production despite the hypoglycaemic TME, and, hence, sustained immunosuppression [46].
2.4. PD-1 and CTLA-4 Signalling and the Effects of Immune Checkpoint Blockade on Metabolic Pathways
PD-1 and CTLA-4 are immune checkpoints that serve as negative regulators of T cell function [47]. Signalling via PD-1 limits T cell activation, preventing excess inflammation and tissue damage [48]. This regulatory function is hijacked by cancer cells that upregulate the ligands PD-L1 and PD-L2 on their surface to dampen anti-tumour immunity [49]. PD-1 contains two intracellular tyrosine motifs that, when engaged by its ligands, result in phosphorylation of tyrosine residues, leading to recruitment of protein tyrosine phosphatases (PTPs) such as SHP2 [49]. PTPs antagonise positive signals from the TCR and CD28, hence antagonising downstream pathways including PI3K/Akt, Ras, ERK, Vav, and PLCγ, which are key pathways required for metabolic reprogramming of activated T cells [50][51][52].
Cancer therapy has entered the ‘immunotherapy era’ with anti-PD-1/PD-L1 and anti-CTLA-4 antibodies being incorporated into the treatment for melanoma, triple-negative breast cancer (TNBC), NSCLC, and metastatic renal cell carcinoma (RCC), among others. Such immune checkpoint blockade (ICB) therapy, aimed at reversing the immune suppression caused by tumour cells, also shapes the TME by affecting tumour metabolism [53]. Ligation of PD-1 on activated T cells impair glycolysis or amino acid metabolism [54], while PD-1 blockade upregulates GLUT1 to restore glucose uptake, promoting glycolysis in effector T cells [11]. Furthermore, PD-1 promotes FAO of endogenous lipids by increasing expression of CTP1A and upregulating lipolysis [54]. ROS generation by activators of mTOR, AMPK, and PGC-1α were found to synergise with PD-1 blockade [55]. Taken together, this strengthens the role of combining PD-1 blocking therapies with metabolism-based therapies for more efficacious anti-tumour immunity.
Ligation of CTLA-4 also leads to similar inhibition of key metabolic reprogramming. However, CTLA-4 signalling on T cells inhibits glycolysis without augmenting FAO [54]. This suggests diverging roles of these two immune checkpoints: CTLA-4 sustains the metabolic profile of non-activated cells, while PD-1 functions to dampen metabolic reprogramming in activated cells [54]. Regardless, the function of PD-1 and CTLA-4 in antagonising key metabolic pathways in T cells are mechanisms tumours used to limit anti-tumour immunity, and provides an explanation for the capacity of T cells to be metabolically invigorated by ICB.
2.5. Resistance to Immunotherapies
However, in reality, only a small proportion of patients respond well to ICB [56]. Studies have uncovered several reasons for this gap, including poor tumour immunogenicity, tumour editing, and lack of sufficient tumour-infiltrating T cells in a ‘cold’ tumour [57][58].
The altered metabolism in cancer cells is often associated with dysregulated expression of key metabolic enzymes. The aberrantly expressed enzymes have pleiotropic effects that contribute to immunosuppression, limiting the effectiveness of immunotherapies. For example, many cancers display MYC-dependent upregulation of the alternatively spliced PKM2 enzyme as a mechanism to enhance aerobic glycolysis [59]. Furthermore, independent of its enzymatic action on glycolysis, PKM2 promotes the expression of PD-L1 on tumour surfaces and, hence, promotes immune suppression [60]. PKM2 activity also aids in recruitment of MDSCs, and is associated with increased metastasis and poor prognosis in HCC [61].
Tumour metabolism also limits the effectiveness of immunotherapies by affecting the tumour mutation rate and antigenicity [62]. Metabolism is tightly linked to DNA repair through chromatin remodelling, epigenetic modifications, and regulation of the redox status [63]. Altered tumour metabolism can promote chromatin remodelling and epigenetic modifications in multiple ways, such as by supplying acetyl and methyl groups and producing metabolites that act as key cofactors or inhibitors of epigenetic enzymes, such as α-KG, succinate, fumarate, and 2-hydroxyglutarate [64]. Furthermore, the enhanced nucleotide biosynthesis in tumours promotes DNA repair [9]. Taken together, these processes lead to a reduced mutation rate and, hence, reduced tumour antigenicity, thereby limiting the effectiveness of immunotherapies.
Finally, CAFs in the TME may also contribute to immunotherapy resistance by several mechanisms. Firstly, the release of immunosuppressive cytokines TGF-β and IL-6 by CAFs lead to reduced proliferation and trafficking capacity of antigen-presenting DCs, thereby impairing T cell priming against tumour antigens [62][65]. CAFs also directly upregulate immune checkpoint ligands on their surface, including PD-L1 and PD-L2 [66][67]. Next, CAFs impair T cell migration to the tumour bed. Through tight regulation of the local chemokine gradient, CAFs limit T cell attraction to the TME [68][69]. CAFs also impair T cell access to the tumour, directly via inhibition of T cell migration through a TGF-β-dependent gene programme [70] as well as indirectly by altering the composition of the ECM, creating a denser ECM network, which functions as a physical barrier to T cell infiltration [71][72].
3. Therapeutic Opportunities Against Altered CCM
With greater understanding of dysregulated cancer metabolism and the metabolic interplay of cancer cells with the tumour immune microenvironment, therapies can be developed to target these processes and overcome therapeutic resistance. Metabolic pathways that are being targeted include glycolysis, OXPHOS and glutaminolysis. Various trials are also being undertaken for metabolic inhibition plus immunotherapy combinations. The anti-tumour activity of immune checkpoint inhibition may be enhanced by metabolic modulation of the TME. For instance, promising preclinical data combining the glutaminase inhibitor CB-839 with immune checkpoint blockade (ICB) therapy has led to an ongoing phase I/II study of CB-839 in combination with nivolumab in immunogenic tumours including melanoma, RCC, and NSCLC (NCT02771626) (Table 1) [73]. Targeting the IDO-dependent tryptophan degradation, IDO inhibitors also reduces the production of immunosuppressive kynurenine. Multiple phase I/II trials showed encouraging results with small molecule inhibitors of IDO1, such as epacadostat, with improved responses to anti-PD-1 therapy (Table 1). However, recent results from ECHO-301, the first large phase III trial to evaluate the efficacy of epacadostat in combination with pembrolizumab in advanced melanoma, showed no indication that epacadostat provided an additional benefit. Thus, the current usefulness of IDO1 inhibition to enhance anti-PD-1 therapy remains to be seen. Other IDO1 inhibitors are being developed and, in earlier phase trials, including navoximod, currently being tested in a phase Ib trial for solid tumours in combination with atezolizumab [74].
Table 1. Metabolic inhibitors in combination with immunotherapy.
| Targeted Metabolism | Metabolic Inhibitor | Immunotherapy | Preclinical Data | Clinical Data |
|---|---|---|---|---|
| Glutaminolysis | CB-839 | Anti-PD-1, anti-PD-L1 | Colon [73] | |
| Nivolumab | Phase I/II—melanoma, RCC, NSCLC (NCT02771626) | |||
| Pembrolizumab + carboplatin + pemetrexed | Phase II—NSCLC (NCT04265534) | |||
| JHU083 | Anti-PD-1 | Lymphoma, colon, melanoma [21] | ||
| Amino acid metabolism | CB-1158/INCB001158 (Arg1 inhibitor) | Anti-PD-1 Pembrolizumab Daratumumab |
Solid tumours [75] | Phase I/II—solid tumours (NCT02903914) Phase I/II—MM (NCT03837509) |
| Epacadostat/INCB024360 (IDO1 inhibitor) |
Checkpoint inhibitors (various) | Phase I/II—solid tumours (multiple clinical trials) | ||
| Pembrolizumab | Phase III—melanoma (NCT02752074) [74] Phase III—melanoma, urothelial carcinoma, HNSCC (Keynote-ECHO trials: NCT02752074, NCT03361865, NCT03374488, NCT03358472) |
|||
| Navoximod/GDC-0919 (IDO1 inhibitor) |
Atezolizumab | Phase Ib—solid tumours (NCT02471846, NCT02048709) | ||
| Other | CPI-444/ciforadenant (A2AR antagonist) |
Atezolizumab | Phase I—RCC, prostate (NCT02655822) Phase I/II—NSCLC (NCT03337698) |
|
| Daratumumab | Phase I—MM (NCT04280328) |
Indeed, the dynamic metabolic crosstalk between cancer cells and the TME, in particular the immune system, adds further layers of complexity to cancer therapy. Various immunosuppressive strategies are exerted by the tumour cells on the immune system, partially accounting for the reduced effectiveness of ICB therapies [56]. Nevertheless, important strides have been made toward the clinical application of metabolic inhibition to synergize with immune anti-tumour activity.
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674, doi:10.1016/J.CELL.2011.02.013.
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314, doi:10.1126/science.123.3191.309.
- Liberti, M. V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends in biochemical sciences 2016, 41, 211, doi:10.1016/J.TIBS.2015.12.001.
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clinical Cancer Research 2018, 24, 2482–2490, doi:10.1158/1078-0432.CCR-17-3070.
- Haq, R.; Shoag, J.; Andreu-Perez, P.; Yokoyama, S.; Edelman, H.; Rowe, G.C.; Frederick, D.T.; Hurley, A.D.; Nellore, A.; Kung, A.L.; et al. Oncogenic BRAF Regulates Oxidative Metabolism via PGC1α and MITF. Cancer Cell 2013, 23, 302–315, doi:10.1016/J.CCR.2013.02.003.
- Hirpara, J.; Eu, J.Q.; Tan, J.K.M.; Wong, A.L.; Clement, M.-V.; Kong, L.R.; Ohi, N.; Tsunoda, T.; Qu, J.; Goh, B.C. Metabolic reprogramming of oncogene-addicted cancer cells to OXPHOS as a mechanism of drug resistance. Redox Biology 2019, 25, 101076, doi:10.1016/J.REDOX.2018.101076.
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Science advances 2016, 2, e1600200, doi:10.1126/sciadv.1600200.
- Schwörer, S.; Vardhana, S.A.; Thompson, C.B. Cancer Metabolism Drives a Stromal Regenerative Response. Cell metabolism 2019, 29, 576–591, doi:10.1016/j.cmet.2019.01.015.
- Coleman, M.F.; Cozzo, A.J.; Pfeil, A.J.; Etigunta, S.K.; Hursting, S.D. Cell Intrinsic and Systemic Metabolism in Tumor Immunity and Immunotherapy. Cancers 2020, 12, 852, doi:10.3390/cancers12040852.
- Cairns, R.A.; Mak, T.W. Fire and water: Tumor cell adaptation to metabolic conditions. Experimental Cell Research 2017, 356, 204–208, doi:10.1016/J.YEXCR.2017.04.029.
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.W.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241, doi:10.1016/J.CELL.2015.08.016.
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death & Disease 2015, 6, e1792–e1792, doi:10.1038/cddis.2015.162.
- Zhang, Y.; Kurupati, R.; Liu, L.; Zhou, X.Y.; Zhang, G.; Hudaihed, A.; Filisio, F.; Giles-Davis, W.; Xu, X.; Karakousis, G.C.; et al. Enhancing CD8+ T Cell Fatty Acid Catabolism within a Metabolically Challenging Tumor Microenvironment Increases the Efficacy of Melanoma Immunotherapy. Cancer cell 2017, 32, 377-391.e9, doi:10.1016/j.ccell.2017.08.004.
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nature Reviews Immunology 2015, 15, 486–499, doi:10.1038/nri3862.
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; Yu, X.; Huseni, M.; Yang, Y.; Park, S.; Javinal, V.; Chiu, H.; Irving, B.; et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell 2014, 26, 923–937, doi:10.1016/j.ccell.2014.10.018.
- Chauvin, J.-M.; Pagliano, O.; Fourcade, J.; Sun, Z.; Wang, H.; Sander, C.; Kirkwood, J.M.; Chen, T.T.; Maurer, M.; Korman, A.J.; et al. TIGIT and PD-1 impair tumor antigen-specific CD8+ T cells in melanoma patients. J. Clin. Invest. 2015, 125, 2046–2058, doi:10.1172/JCI80445.
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen–specific CD8+ T cell dysfunction in melanoma patients. J Exp Med 2010, 207, 2175–2186, doi:10.1084/jem.20100637.
- Peng, M.; Yin, N.; Chhangawala, S.; Xu, K.; Leslie, C.S.; Li, M.O. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 2016, 354, 481–484, doi:10.1126/SCIENCE.AAF6284.
- Nakaya, M.; Xiao, Y.; Zhou, X.; Chang, J.-H.; Chang, M.; Cheng, X.; Blonska, M.; Lin, X.; Sun, S.-C. Inflammatory T Cell Responses Rely on Amino Acid Transporter ASCT2 Facilitation of Glutamine Uptake and mTORC1 Kinase Activation. Immunity 2014, 40, 692–705, doi:10.1016/j.immuni.2014.04.007.
- Carr, E.L.; Kelman, A.; Wu, G.S.; Gopaul, R.; Senkevitch, E.; Aghvanyan, A.; Turay, A.M.; Frauwirth, K.A. Glutamine Uptake and Metabolism Are Coordinately Regulated by ERK/MAPK during T Lymphocyte Activation. The Journal of Immunology 2010, 185, 1037–1044, doi:10.4049/JIMMUNOL.0903586.
- Leone, R.D.; Zhao, L.; Englert, J.M.; Sun, I.-M.; Oh, M.-H.; Sun, I.-H.; Arwood, M.L.; Bettencourt, I.A.; Patel, C.H.; Wen, J.; et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science (New York, N.Y.) 2019, 366, 1013–1021, doi:10.1126/science.aav2588.
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 2016, 167, 829-842.e13, doi:10.1016/J.CELL.2016.09.031.
- Mándi, Y.; Vécsei, L. The kynurenine system and immunoregulation. Journal of Neural Transmission 2012, 119, 197–209, doi:10.1007/s00702-011-0681-y.
- O’Sullivan, D.; van der Windt, G.J.W.; Huang, S.C.-C.; Curtis, J.D.; Chang, C.-H.; Buck, M.D.; Qiu, J.; Smith, A.M.; Lam, W.Y.; DiPlato, L.M.; et al. Memory CD8+ T Cells Use Cell-Intrinsic Lipolysis to Support the Metabolic Programming Necessary for Development. Immunity 2014, 41, 75–88, doi:10.1016/J.IMMUNI.2014.06.005.
- Klysz, D.; Tai, X.; Robert, P.A.; Craveiro, M.; Cretenet, G.; Oburoglu, L.; Mongellaz, C.; Floess, S.; Fritz, V.; Matias, M.I.; et al. Glutamine-dependent α-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Science Signaling 2015, 8, ra97–ra97, doi:10.1126/SCISIGNAL.AAB2610.
- Anderson, K.G.; Stromnes, I.M.; Greenberg, P.D. Obstacles posed by the tumor microenvironment to T cell activity: a case for synergistic therapies. Cancer Cell 2017, 31, 311–325, doi:10.1016/j.ccell.2017.02.008.
- Angelin, A.; Gil-de-Gómez, L.; Dahiya, S.; Jiao, J.; Guo, L.; Levine, M.H.; Wang, Z.; Quinn, W.J.; Kopinski, P.K.; Wang, L.; et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell metabolism 2017, 25, 1282-1293.e7, doi:10.1016/j.cmet.2016.12.018.
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell–derived lactic acid on human T cells. Blood 2007, 109, 3812–3819, doi:10.1182/blood-2006-07-035972.
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metabolism 2016, 24, 657–671, doi:10.1016/J.CMET.2016.08.011.
- Waickman, A.T.; Powell, J.D. mTOR, metabolism, and the regulation of T-cell differentiation and function. Immunological Reviews 2012, 249, 43–58, doi:10.1111/j.1600-065X.2012.01152.x.
- Choi, S.Y.C.; Collins, C.C.; Gout, P.W.; Wang, Y. Cancer-generated lactic acid: a regulatory, immunosuppressive metabolite? The Journal of Pathology 2013, 230, 350–355, doi:10.1002/path.4218.
- Ohta, A.; Gorelik, E.; Prasad, S.J.; Ronchese, F.; Lukashev, D.; Wong, M.K.K.; Huang, X.; Caldwell, S.; Liu, K.; Smith, P.; et al. A2A adenosine receptor protects tumors from antitumor T cells. Proceedings of the National Academy of Sciences 2006, 103, 13132–13137, doi:10.1073/pnas.0605251103.
- Fallarino, F.; Grohmann, U.; Vacca, C.; Orabona, C.; Spreca, A.; Fioretti, M.C.; Puccetti, P. T Cell Apoptosis by Kynurenines. In; Springer, Boston, MA, 2003; pp. 183–190.
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.-I.; Cheng, P.; Cho, H.-I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. The Journal of experimental medicine 2010, 207, 2439–53, doi:10.1084/jem.20100587.
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563, doi:10.1038/nature13490.
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. Journal of Experimental Medicine 2014, 211, 781–790, doi:10.1084/jem.20131916.
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends in Immunology 2016, 37, 208–220, doi:10.1016/J.IT.2016.01.004.
- Dietl, K.; Renner, K.; Dettmer, K.; Timischl, B.; Eberhart, K.; Dorn, C.; Hellerbrand, C.; Kastenberger, M.; Kunz-Schughart, L.A.; Oefner, P.J.; et al. Lactic Acid and Acidification Inhibit TNF Secretion and Glycolysis of Human Monocytes. The Journal of Immunology 2010, 184, 1200–1209, doi:10.4049/JIMMUNOL.0902584.
- Zelenay, S.; van der Veen, A.G.; Böttcher, J.P.; Snelgrove, K.J.; Rogers, N.; Acton, S.E.; Chakravarty, P.; Girotti, M.R.; Marais, R.; Quezada, S.A.; et al. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 2015, 162, 1257–1270, doi:10.1016/J.CELL.2015.08.015.
- Giovanelli, P.; Sandoval, T.A.; Cubillos-Ruiz, J.R. Dendritic Cell Metabolism and Function in Tumors. Trends in immunology 2019, 40, 699–718, doi:10.1016/j.it.2019.06.004.
- Krawczyk, C.M.; Holowka, T.; Sun, J.; Blagih, J.; Amiel, E.; DeBerardinis, R.J.; Cross, J.R.; Jung, E.; Thompson, C.B.; Jones, R.G.; et al. Toll-like receptor–induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 2010, 115, 4742–4749, doi:10.1182/blood-2009-10-249540.
- Zhao, F.; Xiao, C.; Evans, K.S.; Theivanthiran, T.; DeVito, N.; Holtzhausen, A.; Liu, J.; Liu, X.; Boczkowski, D.; Nair, S.; et al. Paracrine Wnt5a-β-Catenin Signaling Triggers a Metabolic Program that Drives Dendritic Cell Tolerization. Immunity 2018, 48, 147-160.e7, doi:10.1016/J.IMMUNI.2017.12.004.
- Belladonna, M.L.; Orabona, C.; Grohmann, U.; Puccetti, P. TGF-β and kynurenines as the key to infectious tolerance. Trends in Molecular Medicine 2009, 15, 41–49, doi:10.1016/J.MOLMED.2008.11.006.
- Eibinger, G.; Fauler, G.; Bernhart, E.; Frank, S.; Hammer, A.; Wintersperger, A.; Eder, H.; Heinemann, A.; Mischel, P.S.; Malle, E.; et al. On the role of 25-hydroxycholesterol synthesis by glioblastoma cell lines. Implications for chemotactic monocyte recruitment. Experimental Cell Research 2013, 319, 1828–1838, doi:10.1016/J.YEXCR.2013.03.025.
- Assmann, N.; O’Brien, K.L.; Donnelly, R.P.; Dyck, L.; Zaiatz-Bittencourt, V.; Loftus, R.M.; Heinrich, P.; Oefner, P.J.; Lynch, L.; Gardiner, C.M.; et al. Srebp-controlled glucose metabolism is essential for NK cell functional responses. Nature Immunology 2017, 18, 1197–1206, doi:10.1038/ni.3838.
- Rice, C.M.; Davies, L.C.; Subleski, J.J.; Maio, N.; Gonzalez-Cotto, M.; Andrews, C.; Patel, N.L.; Palmieri, E.M.; Weiss, J.M.; Lee, J.-M.; et al. Tumour-elicited neutrophils engage mitochondrial metabolism to circumvent nutrient limitations and maintain immune suppression. Nature Communications 2018, 9, 5099–5099, doi:10.1038/s41467-018-07505-2.
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways. American Journal of Clinical Oncology 2016, 39, 98–106, doi:10.1097/COC.0000000000000239.
- Frebel, H.; Nindl, V.; Schuepbach, R.A.; Braunschweiler, T.; Richter, K.; Vogel, J.; Wagner, C.A.; Loffing-Cueni, D.; Kurrer, M.; Ludewig, B.; et al. Programmed death 1 protects from fatal circulatory failure during systemic virus infection of mice. The Journal of Experimental Medicine 2012, 209, 2485–2499, doi:10.1084/jem.20121015.
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nature Reviews Immunology 2018, 18, 153–167, doi:10.1038/nri.2017.108.
- Hui, E.; Cheung, J.; J, Z.; X, S.; MJ, T.; HA, W.; DK, S.; J, H.; JM, K.; I, M.; et al. T Cell Costimulatory Receptor CD28 Is a Primary Target for PD-1-mediated Inhibition. Science (New York, N.Y.) 2017, 355, doi:10.1126/SCIENCE.AAF1292.
- Parry, R.; Chemnitz, J.; KA, F.; AR, L.; I, B.; SV, K.; PS, L.; CB, T.; JL, R. CTLA-4 and PD-1 Receptors Inhibit T-cell Activation by Distinct Mechanisms. Molecular and cellular biology 2005, 25, doi:10.1128/MCB.25.21.9543-9553.2005.
- Yokosuka, T.; Takamatsu, M.; W, K.-I.; A, H.-T.; M, A.; T, S. Programmed Cell Death 1 Forms Negative Costimulatory Microclusters That Directly Inhibit T Cell Receptor Signaling by Recruiting Phosphatase SHP2. The Journal of experimental medicine 2012, 209, doi:10.1084/JEM.20112741.
- Chen, L.; Han, X. Anti–PD-1/PD-L1 therapy of human cancer: past, present, and future. The Journal of Clinical Investigation 2015, 125, 3384–3391, doi:10.1172/JCI80011.
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nature Communications 2015, 6, 6692, doi:10.1038/ncomms7692.
- Chamoto, K.; Chowdhury, P.S.; Kumar, A.; Sonomura, K.; Matsuda, F.; Fagarasan, S.; Honjo, T. Mitochondrial activation chemicals synergize with surface receptor PD-1 blockade for T cell-dependent antitumor activity. Proceedings of the National Academy of Sciences 2017, 114, E761–E770, doi:10.1073/PNAS.1620433114.
- Sambi, M.; Bagheri, L.; Szewczuk, M.R. Current Challenges in Cancer Immunotherapy: Multimodal Approaches to Improve Efficacy and Patient Response Rates. Journal of oncology 2019, 2019, 4508794, doi:10.1155/2019/4508794.
- Whiteside, T.L.; Demaria, S.; Rodriguez-Ruiz, M.E.; Zarour, H.M.; Melero, I. Emerging Opportunities and Challenges in Cancer Immunotherapy. Clinical cancer research : an official journal of the American Association for Cancer Research 2016, 22, 1845–55, doi:10.1158/1078-0432.CCR-16-0049.
- Emens, L.A.; Ascierto, P.A.; Darcy, P.K.; Demaria, S.; Eggermont, A.M.M.; Redmond, W.L.; Seliger, B.; Marincola, F.M. Cancer immunotherapy: Opportunities and challenges in the rapidly evolving clinical landscape. European Journal of Cancer 2017, 81, 116–129, doi:10.1016/J.EJCA.2017.01.035.
- David, C.J.; Chen, M.; Assanah, M.; Canoll, P.; Manley, J.L. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 2010, 463, 364–368, doi:10.1038/nature08697.
- Palsson-McDermott, E.M.; Dyck, L.; Zasłona, Z.; Menon, D.; McGettrick, A.F.; Mills, K.H.G.; O’Neill, L.A. Pyruvate Kinase M2 Is Required for the Expression of the Immune Checkpoint PD-L1 in Immune Cells and Tumors. Frontiers in Immunology 2017, 8, 1300, doi:10.3389/fimmu.2017.01300.
- Liu, W.-R.; Tian, M.-X.; Yang, L.-X.; Lin, Y.-L.; Jin, L.; Ding, Z.-B.; Shen, Y.-H.; Peng, Y.-F.; Gao, D.-M.; Zhou, J.; et al. PKM2 promotes metastasis by recruiting myeloid-derived suppressor cells and indicates poor prognosis for hepatocellular carcinoma. Oncotarget 2015, 6, 846–61, doi:10.18632/oncotarget.2749.
- van Elsas, M.J.; van Hall, T.; van der Burg, S.H. Future Challenges in Cancer Resistance to Immunotherapy. Cancers 2020, 12, 935–935, doi:10.3390/cancers12040935.
- Turgeon, M.-O.; Perry, N.J.S.; Poulogiannis, G. DNA Damage, Repair, and Cancer Metabolism. Frontiers in Oncology 2018, 8, 15, doi:10.3389/fonc.2018.00015.
- Kinnaird, A.; Zhao, S.; Wellen, K.E.; Michelakis, E.D. Metabolic control of epigenetics in cancer. Nature Reviews Cancer 2016, 16, 694–707.
- Harryvan, T.J.; Verdegaal, E.M.E.; Hardwick, J.C.H.; Hawinkels, L.J.A.C.; van der Burg, S.H. Targeting of the Cancer-Associated Fibroblast—T-Cell Axis in Solid Malignancies. Journal of Clinical Medicine 2019, 8, 1989, doi:10.3390/jcm8111989.
- Pinchuk, I. V; Saada, J.I.; Beswick, E.J.; Boya, G.; Qiu, S.M.; Mifflin, R.C.; Raju, G.S.; Reyes, V.E.; Powell, D.W. PD-1 ligand expression by human colonic myofibroblasts/fibroblasts regulates CD4+ T-cell activity. Gastroenterology 2008, 135, 1228–1237, 1237.e1–2, doi:10.1053/j.gastro.2008.07.016.
- Lakins, M.A.; Ghorani, E.; Munir, H.; Martins, C.P.; Shields, J.D. Cancer-associated fibroblasts induce antigen-specific deletion of CD8 + T Cells to protect tumour cells. Nature Communications 2018, 9, 948, doi:10.1038/s41467-018-03347-0.
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel III, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548, doi:10.1038/nature25501.
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543, doi:10.1038/nature25492.
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science (New York, N.Y.) 2006, 313, 1960–4, doi:10.1126/science.1129139.
- Salmon, H.; Franciszkiewicz, K.; Damotte, D.; Dieu-Nosjean, M.-C.; Validire, P.; Trautmann, A.; Mami-Chouaib, F.; Donnadieu, E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. The Journal of clinical investigation 2012, 122, 899–910, doi:10.1172/JCI45817.
- Chakravarthy, A.; Khan, L.; Bensler, N.P.; Bose, P.; De Carvalho, D.D. TGF-β-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nature Communications 2018, 9, 4692, doi:10.1038/s41467-018-06654-8.
- Gross, M.; Chen, J.; Englert, J.; Janes, J.; Leone, R.; MacKinnon, A.; Parlati, F.; Rodriquez, M.; Shwonek, P.; Powell, J. Abstract 2329: Glutaminase inhibition with CB-839 enhances anti-tumor activity of PD-1 and PD-L1 antibodies by overcoming a metabolic checkpoint blocking T cell activation. In Proceedings of the 107th Annual Meeting of the American Association for Cancer Research; 2016 Apr 16-20; New Orleans, LA; American Association for Cancer Research, 2016; Vol. 76, pp. 2329–2329.
- Long, G. V; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.-J.; Kim, T.M.; et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. The Lancet Oncology 2019, 20, 1083–1097, doi:10.1016/S1470-2045(19)30274-8.
- Steggerda, S.M.; Bennett, M.K.; Chen, J.; Emberley, E.; Huang, T.; Janes, J.R.; Li, W.; MacKinnon, A.L.; Makkouk, A.; Marguier, G.; et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. Journal for ImmunoTherapy of Cancer 2017, 5, 101, doi:10.1186/s40425-017-0308-4.

