Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Qingzhong Wang | -- | 2354 | 2022-11-13 12:25:38 | | | |
| 2 | Beatrix Zheng | Meta information modification | 2354 | 2022-11-14 03:08:28 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Peng, S.; Wang, H.; Wang, Z.; Wang, Q. Antiviral Agents Targeting Viral Polymerases. Encyclopedia. Available online: https://encyclopedia.pub/entry/34243 (accessed on 09 August 2026).
Peng S, Wang H, Wang Z, Wang Q. Antiviral Agents Targeting Viral Polymerases. Encyclopedia. Available at: https://encyclopedia.pub/entry/34243. Accessed August 09, 2026.
Peng, Siqi, Huizhen Wang, Zhengtao Wang, Qingzhong Wang. "Antiviral Agents Targeting Viral Polymerases" Encyclopedia, https://encyclopedia.pub/entry/34243 (accessed August 09, 2026).
Peng, S., Wang, H., Wang, Z., & Wang, Q. (2022, November 13). Antiviral Agents Targeting Viral Polymerases. In Encyclopedia. https://encyclopedia.pub/entry/34243
Peng, Siqi, et al. "Antiviral Agents Targeting Viral Polymerases." Encyclopedia. Web. 13 November, 2022.
Copy Citation
Viral DNA and RNA polymerases are two kinds of very important enzymes that synthesize the genetic materials of the virus itself, and they have become extremely favorable targets for the development of antiviral drugs because of their relatively conserved characteristics. There are many similarities in the structure and function of different viral polymerases, so inhibitors designed for a certain viral polymerase have acted as effective universal inhibitors on other types of viruses.
viral DNA and RNA polymerase
antiviral drugs
inhibitors
1. Viral DNA/RNA Synthases Inhibitors
After the virus enters the host cell, the host cell is used to synthesize the virus’ own nucleic acid and protein [1]. Most of the antiviral drugs that are clinically used act on the viral replication stage [2].
1.1. Influenza Virus RNA Polymerase Inhibitor
The replication for most viral RdRp is processed within the host cells. Influenza virus RdRp consists of three subunits encoded by the virus: PB1, PB2, and PA [3]. The naked viral genomic RNA must be combined with nucleic proteins to form a complex that serves as a template to initiate viral genome replication and transcription by RdRp. Studies have shown that the three subunits of influenza virus RdRp can enter the nucleus individually [4][5][6]. First, RdRp is transported by the nuclear localization signal (NLS) of the nucleoprotein (NP) into the host’s nucleus for assembly [7]. After assembly, the replication process of the influenza virus’ genetic material begins. Since the influenza virus cannot produce 5’-cap primers on its own, its PB2 subunit captures the 5’-cap structure of host cell RNA through a “cap capture” mechanism [8]. After replication is completed, the product is exported through a separate channel for viral mRNA synthesis [9]. Different types of inhibitors targeting the PB1, PB2, and PA subunits are discussed in detail below [10][11].
1.1.1. PA Inhibitors
At present, the PA inhibitor baloxavir marboxil, jointly developed by Shionogi and Roche, is used for the treatment of influenza A and B in individuals over the age of 12 [12]. Since there is no similar mechanism and corresponding protease in host cells, baloxavir acts as a novel CAP-dependent nucleic acid endonuclease inhibitor, and can selectively block the transcription process of the influenza virus without affecting host cells [13][14]. The results of clinical trials show that baloxavir can significantly improve the time of symptom relief and that this drug is well-tolerated [15].
1.1.2. PB1 Inhibitors
Among the PB1 inhibitors, ribavirin and favipiravir have entered the clinical research stage due to their good antiviral activity [16]. Although both compounds contain bases in their structures, their antiviral mechanisms are not the same [17]. Ribavirin is a broad-spectrum antiviral drug widely accepted as a competitive inhibitor of host monophosphate dehydrogenase (IMPDH) [18]. IMPDH can catalyze the conversion of guanosine monophosphate (GMP) to guanine triphosphate (GTP), and inhibition of this enzyme will reduce the content of GTP in cells, resulting in an imbalance in nucleotide concentration, thereby inhibiting viral protein synthesis and exerting an antiviral effect [19]. Favipiravir belongs to a class of purine analogs that need to be rapidly converted into triphosphate form in vivo, and it exerts an antiviral effect by simulating competitive inhibition of RNA polymerase activity by GTP [20][21]. At the same time, it can also be incorporated into viral genes and exert antiviral effects by inducing lethal mutations [22]. In addition to being anti-influenza virus, favipiravir is effective against a variety of RNA viruses such as Lassa fever virus, Rift Valley fever virus, Hantavirus, Flaviviridae, West Nile virus, Zika virus, Chikungunya virus, Ebola virus, etc [20].
1.1.3. PB2 Inhibitors
Pimodivir, also known as VX-787, is a typical representative of the PB2 cap-binding domain inhibitors [23]. Based on phase III clinical study data, pimodivir did not show better efficacy than the existing standard compound, so Janssen decided to stop its clinical development as an anti-influenza A virus drug [24].
1.1.4. Protein–Protein Interaction Inhibitors
The three subunits of RdRp are non-covalently combined into a functional complex, so blocking the interaction between the subunits can effectively inhibit the activity of RdRp [25]. The polymerase inhibitors developed based on this mechanism are called protein–protein interaction inhibitors (PPI inhibitors) [26]. At present, the most commonly studied PPI inhibitors are PA-PB1 inhibitors. Massari et al. (2013) found that cycloheptathiophene-3-carboxamide compounds had weak PA-PB1 inhibitory activity but no antiviral activity through ELISA experiments, and further modified cycloheptathiophene-3-carboxamide compounds by considering their structure–activity relationship. They also synthesized 35 compounds, of which 1 and 2 had the strongest activity and whose structures are shown in Figure 1; their IC50 values were 32 µmol/L and 35 µmol/L, respectively, and they had no cytotoxicity at the concentration of 250 µmol/L. These two compounds became potential PPI inhibitors [27].
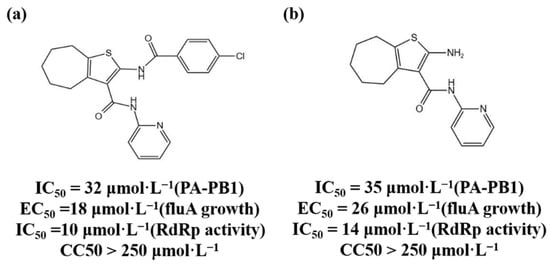
Figure 1. (a,b) Chemical structures of Protein–protein interaction inhibitors and their antiviral activities.
1.2. HCV-related Inhibitors
The NS5B polymerase encoded by the non-structural gene NS5B is an enzyme of RdRp [28]. Its structure has the “right-hand” conformation of a typical RNA polymerase. The “palm subdomain“ is the central part of the polymerase and is the catalytic center of genome replication; the “fingers subdomain“ is responsible for capturing the nucleotide triphosphates required for replication; and the “thumb subdomain“ coordinates the initiation and elongation of RNA replication [29]. HCV replicates by using the viral genome’s single-stranded RNA as a template and joining the replicon created by the NS5B polymerase [30]. Because human cells lack similar enzymes, NS5B polymerase is a promising antiviral drug target. HCV NS5B polymerase inhibitors are divided into two categories: NIs and non-nucleoside inhibitors (NNIs) [31].
1.2.1. NIs
Sofosbuvir is a uridine analog prodrug, which can specifically inhibit HCV NS5B polymerase activity and is used for antiviral therapy in patients with chronic hepatitis C. The mechanism of action involves sofosbuvir being converted into the active uridine triphosphate under the action of intracellular phosphokinase, which competes with intracellular nucleotide phosphate as a substrate for NS5B polymerase and is incorporated into newly synthesized RNA chain, terminating the elongation of the HCV RNA chain, thereby inhibiting the replication of HCV [32]. Sofosbuvir has no inhibitory activity on human DNA and RNA polymerase, nor on mitochondrial RNA polymerase, so it has strong specificity [33]. Meanwhile, Sofosbuvir is a “pan-genotype” anti-HCV drug which not only inhibits HCV genotype 1 disease but is also effective against other genotypes of HCV infection.
1.2.2. NNIs
NNIs bind to the allosteric site of NS5B polymerase, resulting in a change in enzyme conformation, thereby inhibiting the activity of NS5B polymerase and exerting an antiviral effect [34][35]. Compared with NIs, NNIs have a lower genetic barrier and are prone to drug resistance and relapse after drug withdrawal, and cannot have antiviral effects on all genotypes [36]. Among them, dasabuvir sodium hydrate was approved by the European Medicines Agency (EMA) in January 2015 as an NNI-type drug.
1.2.3. NS5A Inhibitors
The HCV nonstructural protein NS5A is one of the components of the viral RNA replication complex [37]. It has not yet been found to have enzymatic activity, but it is essential for HCV RNA replication and is also related to the INF response [38]. NS5A inhibitors may exert anti-HCV effects by inhibiting the hyperphosphorylation of NS5A or altering the subcellular localization of NS5A [39]. Daclatasvir is a representative NS5A inhibitors which has a strong antiviral effect and is a pan-genotype drug [40].
1.3. HIV-1 Reverse Transcriptase Inhibitors
After retroviruses such as HIV-1 enter cells, they first synthesize RNA-DNA complex strands under the action of reverse transcriptase, and then the RNA strands are hydrolyzed to synthesize double-stranded DNA [41]. Under the action of the enzyme, this DNA is embedded into the genome of the host cell and replicated with the transcription and translation machinery of the host cell [42]. HIV-1 RT is the product of HIV-1 pol gene and consists of p51 and p66 subunits; the RT active center is located in the p66 subunit, which has RNA-dependent DNA polymerase activity [41][43]. Its DNA polymerase domain also has a “right-hand” conformation, similar to the RNA virus polymerase. It has four subdomains: the finger, thumb, palm, and linker domains. It is mainly responsible for the synthesis of double-stranded DNA using HIV single-stranded RNA as a template. The finger, thumb, and palm subdomains together constitute the active site, which occurs so that the enzyme is able to locate the template-primer accurately, and the palm domain contains the key site of DNA chain replication and elongation [44]. There are currently two types of reverse transcriptase inhibitors: NRTIs and NNRTIs [45].
1.3.1. NRTIs
NRTIs are analogs of deoxynucleotides, the DNA RT substrates for the synthesis of HIV-1. In vivo, NRTIs are converted into active nucleoside triphosphate derivatives, which compete with the natural deoxynucleoside triphosphate to bind HIV-1 RT to inhibit RT activity or terminate the RNA chain [46][47]. Currently approved NRTIs include zidovudine (AZT), didanosine (ddI), zalcitabine (ddC), stanvudine (d4T), lamivudine (3TC), abacavir (ABC), emtricitabine [(-)FTC], tenofovir (TFV), etc. [48].
1.3.2. NNRTIs
NNRTIs can directly combine with the p66 hydrophobic region of HIV-1 RT to change the conformation of the enzyme protein and inhibit the enzyme’s activity [41][49]. Unlike NRTIs, NNRTIs are not directly incorporated into DNA strands and do not require phosphorylation, and can function in quiescent and activated cells [50]. NNRTIs currently approved for clinical use include nevirapine (NVP), delavirdine (DLV), efavirenz (EFV), etravirine (TMC-125), and rilpivirine (RPV) [51].
1.3.3. HIV-1 Integrase Inhibitors
HIV-1 integrase catalyzes the integration of the HIV-1 virus reverse transcription product cDNA into the human host genome [52]. HIV-1 integrase is encoded by the 3’-end of the viral pol gene and contains 288 amino acid residues [53]. The integrase has 3’ cleavage endonuclease activity or strand transfer activity in vivo [54]. Inhibition of the HIV-1 integration process can be achieved by inhibiting the endonuclease activity or strand transfer activity of the integrase. There are currently four HIV-1 integrase inhibitors approved for clinical mediation, namely raltegravir, elvitegravir, dolutegravir, and bictegravir, and all four drugs act on the chain transfer process [55].
1.3.4. SARS-CoV-2 Enzyme Inhibitors
The structure of the polymerase complex of SARS-CoV-2 includes one NSP12, one NSP7, and two NSP8 subunits, which are required to complete the replication process of the coronavirus RNA [56]. The core catalytic site of the coronavirus polymerase is NSP12, and its RdRp domain is present in the standard “right-hand” conformation, including the three finger, thumb, and palm subdomains [57]. The RdRp associates with additional non-structural proteins to form a replication-transcription complex that carries out RNA synthesis, capping, and proofreading [58]. Remdesivir was originally a drug used for the treatment of hepatitis C and was later shown to be a broad-spectrum antiviral drug with a delayed chain termination mechanism of action [59][60]. Remdesivir is also being evaluated as an anti-coronavirus drug, and is currently the only drug approved by the US FDA for the treatment of patients with COVID-19. Favipiravir was originally used to treat RNA virus infections such as Ebola and influenza, but a randomized clinical trial found that the drug can bind to the RdRp metal catalytic site of SARS-CoV-2 and produce inhibitory activity [61]. Therefore, favipiravir has been urgently approved for the treatment of mild COVID-19 in several countries [62]. At present, the drug has entered phase 3 clinical trials for the treatment of COVID-19 in many countries [63]. Recent studies have found that suramin, an NNI, can effectively inhibit the activity of SARS-CoV-2 RdRp and prevent the virus from entering cells [64]. It has been proved that the anti-COVID-19 mechanism involves two symmetrical suramin molecules binding to RdRp and preventing RNA templates and primers from binding [65]. It binds to the active site and prevents nucleotide triphosphates from entering the catalytic site, thereby inhibiting the growth of SARS-CoV-2 [66].
2. Research and Development Strategies for Novel Antiviral Drugs
Due to the increasing cost of drug research and development, traditional drug random screening strategies and blind optimization of lead compounds consume a lot of resources and time. In recent years, some new research strategies have been developed, which are listed as follows:
2.1. Nucleic Acid Degradation
Ribonuclease targeting chimera (RIBOTAC) technology converts RNA-binding molecules into RNA-degrading molecules to degrade the viral genome by combining RNA-binding molecules with a small molecule and activating ribonuclease L (RNase L) [67]. Haniff et al. (2020) verified through a series of experiments that compound C5 can stabilize the frameshift element and significantly inhibit the frameshift ability of the SARS-CoV-2 frameshift element. At the same time, the structure of compound 39 was modified with the help of RIBOTAC technology, that is, compound 39 was connected to a small molecule that can recruit RNase L through a linking chain of suitable length to achieve the purpose of directional degradation of SARS-CoV-2 mRNA [68].
2.2. Protein Degradation
Targeting protein degradation chimera (proteolysis-targeting chimera, PROTAC) molecules can target degradation of proteins [69]. PROTAC bifunctional small molecules can bring target proteins and E3 ubiquitin ligases closer together, induce ubiquitination of target proteins and subsequent degradation through the ubiquitin-proteasome pathway [70]. Yang et al. (2022) used PROTAC technology to develop anti-HCV molecules, which can induce viral proteasome degradation [71]. The PROTAC molecule combined with the ligand of CRL4CRBN can induce HCV NS3/4A protease degradation, proving that protein degradation contributes to its antiviral activity [72].
2.3. RNA Interference Application Drugs
RNA interference (RNAi) refers to the specific gene expression silencing mediated by double-stranded RNA [73]. ARC-520 is an RNA interference (RNAi)-based drug for the treatment of chronic hepatitis B [74]. It can act on HBV covalently closed circular DNA (cccDNA) transcription to degrade mRNA [75]. Nonclinical toxicology studies in primates found that ARC-520 may be potentially toxic, and temporarily halted clinical trials of this drug [76]. The HBV RNAi drugs currently under development include ARB-1467 (Phase II clinical trial), RG-6004 (Phase I clinical trial), GSK-3389404, and GSK-3228836 (Phase II clinical trial) [77][78][79].
2.4. Capsid Protein Assembly Regulators
The HBV capsid protein assembly regulator can inhibit the replication of the HBV virus by destroying the function of the capsid [80]. The HBV capsid can not only protect the viral genome encapsulated in the capsid, but also promote the reverse transcription of pgRNA to form DNA [81][82]. Heteroaryldihydropyrimidine (HAP) compounds used as HBV capsid assembly modulators (CpAM) can enhance the hydrophobic interaction between adjacent core protein dimers, alter the dimerization angle between the two substances, change their assembly kinetics, and accelerate the degradation of capsid proteins [83][84]. Electron microscopy results show that HAP compounds can promote the formation of large and irregular particles around which misassembled capsids cannot properly wrap pgRNA, and prevent HBV replication [85]. Representative HBV capsid protein assembly regulators include Bay41-4109 (first generation), GIS-4 (second generation, phase 1 clinical trial), and HAP-R10 (third generation, phase I clinical trial) [86][87].
References
- Villanueva, R.A.; Rouillé, Y.; Dubuisson, J. Interactions between virus proteins and host cell membranes during the viral life cycle. Int. Rev. Cytol. 2005, 245, 171–244.
- Razonable, R.R. Antiviral drugs for viruses other than human immunodeficiency virus. Mayo Clin. Proc. 2011, 86, 1009–1026.
- Te Velthuis, A.J.; Fodor, E. Influenza virus RNA polymerase: Insights into the mechanisms of viral RNA synthesis. Nat. Rev. Microbiol. 2016, 14, 479–493.
- Akkina, R.K.; Chambers, T.M.; Londo, D.R.; Nayak, D.P. Intracellular localization of the viral polymerase proteins in cells infected with influenza virus and cells expressing PB1 protein from cloned cDNA. J. Virol. 1987, 61, 2217–2224.
- Jones, I.M.; Reay, P.A.; Philpott, K.L. Nuclear location of all three influenza polymerase proteins and a nuclear signal in polymerase PB2. EMBO J. 1986, 5, 2371–2376.
- Smith, G.L.; Levin, J.Z.; Palese, P.; Moss, B. Synthesis and cellular location of the ten influenza polypeptides individually expressed by recombinant vaccinia viruses. Virology 1987, 160, 336–345.
- Wang, P.; Palese, P.; O’Neill, R.E. The NPI-1/NPI-3 (karyopherin alpha) binding site on the influenza a virus nucleoprotein NP is a nonconventional nuclear localization signal. J. Virol. 1997, 71, 1850–1856.
- Liu, Y.; Qin, K.; Meng, G.; Zhang, J.; Zhou, J.; Zhao, G.; Luo, M.; Zheng, X.J. Structural and functional characterization of K339Tsubstitution identified in the PB2 subunit cap-binding pocket of influenza A virus. J. Biol. Chem. 2013, 288, 11013–11023.
- Reich, S.; Guilligay, D.; Cusack, S. An in vitro fluorescence based study of initiation of RNA synthesis by influenza B polymerase. Nucleic Acids Res. 2017, 45, 3353–3368.
- Kawaguchi, A.; Momose, F.; Nagata, K. Replication-coupled and host factor-mediated encapsidation of the influenza virus genome by viral nucleoprotein. J. Virol. 2011, 85, 6197–6204.
- Hemerka, J.N.; Wang, D.; Weng, Y.; Lu, W.; Kaushik, R.S.; Jin, J.; Harmon, A.F.; Li, F. Detection and characterization of influenza A virus PA-PB2 interaction through a bimolecular fluorescence complementation assay. J. Virol. 2009, 83, 3944–3955.
- Dufrasne, F. Baloxavir Marboxil: An Original New Drug against Influenza. Pharmaceuticals 2021, 15, 28.
- Baker, J.; Block, S.L.; Matharu, B.; Burleigh Macutkiewicz, L.; Wildum, S.; Dimonaco, S.; Collinson, N.; Clinch, B.; Piedra, P.A. Baloxavir Marboxil Single-dose Treatment in Influenza-infected Children: A Randomized, Double-blind, Active Controlled Phase 3 Safety and Efficacy Trial (miniSTONE-2). Pediatr. Infect. Dis. J. 2020, 39, 700.
- Lee, S.M.; Yen, H.L. Targeting the host or the virus: Current and novel concepts for antiviral approaches against influenza virus infection. Antivir. Res. 2012, 96, 391–404.
- Ng, K.E. Xofluza (Baloxavir Marboxil) for the Treatment of Acute Uncomplicated Influenza. Pharm. Ther. 2019, 44, 9–11.
- Agrawal, U.; Raju, R.; Udwadia, Z.F. Favipiravir: A new and emerging antiviral option in COVID-19. Med. J. Armed Forces India 2020, 76, 370–376.
- Kausar, S.; Said Khan, F.; Ishaq Mujeeb Ur Rehman, M.; Akram, M.; Riaz, M.; Rasool, G.; Hamid Khan, A.; Saleem, I.; Shamim, S.; Malik, A. A review: Mechanism of action of antiviral drugs. Int. J. Immunopathol. Pharmacol. 2021, 35, 20587384211002621.
- Markland, W.; McQuaid, T.J.; Jain, J.; Kwong, A.D. Broad-spectrum antiviral activity of the IMP dehydrogenase inhibitor VX-497: A comparison with ribavirin and demonstration of antiviral additivity with alpha interferon. Antimicrob. Agents Chemother. 2000, 44, 859–866.
- Keppeke, G.D.; Chang, C.C.; Peng, M.; Chen, L.-Y.; Lin, W.-C.; Pai, L.-M.; Andrade, L.E.C.; Sung, L.-Y.; Liu, J.-L. IMP/GTP balance modulates cytoophidium assembly and IMPDH activity. Cell Div. 2018, 13, 5.
- Furuta, Y.; Komeno, T.; Nakamura, T. Favipiravir (T-705), a broad spectrum inhibitor of viral RNA polymerase. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017, 93, 449–463.
- Gordon, C.J.; Tchesnokov, E.P.; Feng, J.Y.; Porter, D.P.; Götte, M. The antiviral compound remdesivir potently inhibits RNA-dependent RNA polymerase from Middle East respiratory syndrome coronavirus. J. Biol. Chem. 2020, 295, 4773–4779.
- Pérez-Losada, M.; Arenas, M.; Galán, J.C.; Palero, F.; González-Candelas, F. Recombination in viruses: Mechanisms, methods of study, and evolutionary consequences. Infect. Genet. Evol. 2015, 30, 296–307.
- Patel, M.C.; Chesnokov, A.; Jones, J.; Mishin, V.P.; De La Cruz, J.A.; Nguyen, H.T.; Zanders, N.; Wentworth, D.E.; Davis, T.C.; Gubareva, L.V. Susceptibility of widely diverse influenza a viruses to PB2 polymerase inhibitor pimodivir. Antivir. Res. 2021, 188, 105035.
- King, J.C.; Beigel, J.H.; Ison, M.G.; Rothman, R.E.; Uyeki, T.M.; Walker, R.E.; Neaton, J.D.; Tegeris, J.S.; Zhou, J.A.; Armstrong, K.L.; et al. Clinical Development of Therapeutic Agents for Hospitalized Patients with Influenza: Challenges and Innovations. Open Forum Infect. Dis. 2019, 6, ofz137.
- Mishra, A.; Rathore, A.S. RNA dependent RNA polymerase (RdRp) as a drug target for SARS-CoV2. J. Biomol. Struct. Dyn. 2022, 40, 6039–6051.
- Ran, X.; Gestwicki, J.E. Inhibitors of protein-protein interactions (PPIs): An analysis of scaffold choices and buried surface area. Curr. Opin. Chem. Biol. 2018, 44, 75–86.
- Massari, S.; Nannetti, G.; Goracci, L.; Sancineto, L.; Muratore, G.; Sabatini, S.; Manfroni, G.; Mercorelli, B.; Cecchetti, V.; Facchini, M.; et al. Structural Investigation of Cycloheptathiophene-3-carboxamide Derivatives Targeting Influenza Virus Polymerase Assembly. J. Med. Chem. 2013, 56, 10118–10131.
- Quezada, E.M.; Kane, C.M. The Hepatitis C Virus NS5A Stimulates NS5B During In Vitro RNA Synthesis in a Template Specific Manner. Open Biochem. J. 2009, 3, 39–48.
- Picarazzi, F.; Vicenti, I.; Saladini, F.; Zazzi, M.; Mori, M. Targeting the RdRp of emerging RNA viruses: The structure-based drug design challenge. Molecules 2020, 25, 5695.
- Zhang, C.; Cai, Z.; Kim, Y.C.; Kumar, R.; Yuan, F.; Shi, P.Y.; Kao, C.; Luo, G. Stimulation of hepatitis C virus (HCV) nonstructural protein 3 (NS3) helicase activity by the NS3 protease domain and by HCV RNA-dependent RNA polymerase. J. Virol. 2005, 79, 8687–8697.
- Membreno, F.E.; Lawitz, E.J. The HCV NS5B nucleoside and non-nucleoside inhibitors. Clin. Liver Dis. 2011, 15, 611–626.
- Te, H.S.; Randall, G.; Jensen, D.M. Mechanism of action of ribavirin in the treatment of chronic hepatitis C. Gastroenterol. Hepatol. 2007, 3, 218–225.
- Jin, Z.; Kinkade, A.; Behera, I.; Chaudhuri, S.; Tucker, K.; Dyatkina, N.; Rajwanshi, V.K.; Wang, G.; Jekle, A.; Smith, D.B.; et al. Structure-activity relationship analysis of mitochondrial toxicity caused by antiviral ribonucleoside analogs. Antivir. Res. 2017, 143, 151–161.
- Davis, B.C.; Brown, J.A.; Thorpe, I.F. Allosteric inhibitors have distinct effects, but also common modes of action, in the HCV polymerase. Biophys. J. 2015, 108, 1785–1795.
- Guo, Q.; He, Y.; Lu, H.P. Interrogating the activities of conformational deformed enzyme by single-molecule fluorescence-magnetic tweezers microscopy. Proc. Natl. Acad. Sci. USA 2015, 112, 13904–13909.
- Pawlotsky, J.M. Therapeutic implications of hepatitis C virus resistance to antiviral drugs. Therap. Adv. Gastroenterol. 2009, 2, 205–219.
- Polyak, S.J.; Khabar, K.S.; Paschal, D.M.; Ezelle, H.J.; Duverlie, G.; Barber, G.N.; Levy, D.E.; Mukaida, N.; Gretch, D.R. Hepatitis C virus nonstructural 5A protein induces interleukin-8, leading to partial inhibition of the interferon-induced antiviral response. J. Virol. 2001, 75, 6095–6106.
- Scheel, T.K.; Rice, C.M. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat. Med. 2013, 19, 837–849.
- Kohler, J.J.; Nettles, J.H.; Amblard, F.; Hurwitz, S.J.; Bassit, L.; Stanton, R.A.; Ehteshami, M.; Schinazi, R.F. Approaches to hepatitis C treatment and cure using NS5A inhibitors. Infect. Drug Resist. 2014, 7, 41–56.
- Sundaram, V.; Kowdley, K.V. Dual daclatasvir and sofosbuvir for treatment of genotype 3 chronic hepatitis C virus infection. Expert Rev. Gastroenterol. Hepatol. 2016, 10, 13–20.
- Sarafianos, S.G.; Marchand, B.; Das, K.; Himmel, D.M.; Parniak, M.A.; Hughes, S.H.; Arnold, E. Structure and function of HIV-1 reverse transcriptase: Molecular mechanisms of polymerization and inhibition. J. Mol. Biol. 2009, 385, 693–713.
- Rampersad, S.; Tennant, P. Replication and Expression Strategies of Viruses. Viruses 2018, 2018, 55–82.
- Schultz, S.J.; Champoux, J.J. RNase H activity: Structure, specificity, and function in reverse transcription. Virus Res. 2008, 134, 86–103.
- Li, G.; De Clercq, E. HIV genome-wide protein associations: A review of 30 years of research. Microbiol. Mol. Biol. Rev. 2016, 80, 679–731.
- Rai, M.A.; Pannek, S.; Fichtenbaum, C.J. Emerging reverse transcriptase inhibitors for HIV-1 infection. Expert Opin. Emerg. Drugs 2018, 23, 149–157.
- Holec, A.D.; Mandal, S.; Prathipati, P.K.; Destache, C.J. Nucleotide Reverse Transcriptase Inhibitors: A Thorough Review, Present Status and Future Perspective as HIV Therapeutics. Curr. HIV Res. 2017, 15, 411–421.
- Dilmore, C.R.; DeStefano, J.J. HIV Reverse Transcriptase Pre-Steady-State Kinetic Analysis of Chain Terminators and Translocation Inhibitors Reveals Interactions between Magnesium and Nucleotide 3′-OH. ACS Omega 2021, 6, 14621–14628.
- García-Lerma, J.G.; MacInnes, H.; Bennett, D.; Reid, P.; Nidtha, S.; Weinstock, H.; Kaplan, J.E.; Heneine, W. A novel genetic pathway of human immunodeficiency virus type 1 resistance to stavudine mediated by the K65R mutation. J. Virol. 2003, 77, 5685–5693.
- De Clercq, E. The role of non-nucleoside reverse transcriptase inhibitors (NNRTIs) in the therapy of HIV-1 infection. Antivir. Res. 1998, 38, 153–179.
- Lanz, M.C.; Dibitetto, D.; Smolka, M.B. DNA damage kinase signaling: Checkpoint and repair at 30 years. Embo J. 2019, 38, e101801.
- Usach, I.; Melis, V.; Peris, J.E. Non-nucleoside reverse transcriptase inhibitors: A review on pharmacokinetics, pharmacodynamics, safety and tolerability. J Int. AIDS Soc. 2013, 16, 18567.
- Tasara, T.; Maga, G.; Hottiger, M.O.; Hübscher, U. HIV-1 reverse transcriptase and integrase enzymes physically interact and inhibit each other. FEBS Lett. 2001, 507, 39–44.
- Blanco, J.L.; Varghese, V.; Rhee, S.Y.; Gatell, J.M.; Shafer, R.W. HIV-1 integrase inhibitor resistance and its clinical implications. J. Infect. Dis. 2011, 203, 1204–1214.
- Craigie, R. The molecular biology of HIV integrase. Future Virol. 2012, 7, 679–686.
- Smith, S.J.; Zhao, X.Z.; Passos, D.O.; Lyumkis, D.; Burke, T.R., Jr.; Hughes, S.H. HIV-1 Integrase Inhibitors That Are Active against Drug-Resistant Integrase Mutants. Antimicrob. Agents Chemother. 2020, 64, e00611–e00620.
- Hillen, H.S.; Kokic, G.; Farnung, L.; Dienemann, C.; Tegunov, D.; Cramer, P.J.N. Structure of replicating SARS-CoV-2 polymerase. Nature 2020, 584, 154–156.
- Shannon, A.; Le, N.T.-T.; Selisko, B.; Eydoux, C.; Alvarez, K.; Guillemot, J.-C.; Decroly, E.; Peersen, O.; Ferron, F.; Canard, B. Remdesivir and SARS-CoV-2: Structural requirements at both nsp12 RdRp and nsp14 Exonuclease active-sites. Antivir. Res. 2020, 178, 104793.
- Hillen, H.S. Structure and function of SARS-CoV-2 polymerase. Curr. Opin. Virol. 2021, 48, 82–90.
- Tian, L.; Qiang, T.; Liang, C.; Ren, X.; Jia, M.; Zhang, J.; Li, J.; Wan, M.; YuWen, X.; Li, H.; et al. RNA-dependent RNA polymerase (RdRp) inhibitors: The current landscape and repurposing for the COVID-19 pandemic. Eur. J. Med. Chem. 2021, 213, 113201.
- Eastman, R.T.; Roth, J.S.; Brimacombe, K.R.; Simeonov, A.; Shen, M.; Patnaik, S.; Hall, M.D. Remdesivir: A Review of Its Discovery and Development Leading to Emergency Use Authorization for Treatment of COVID-19. ACS Cent. Sci. 2020, 6, 672–683.
- Maheden, K.; Todd, B.; Gordon, C.J.; Tchesnokov, E.P.; Götte, M. Inhibition of viral RNA-dependent RNA polymerases with clinically relevant nucleotide analogs. Enzymes 2021, 49, 315–354.
- Hassanipour, S.; Arab-Zozani, M.; Amani, B.; Heidarzad, F.; Fathalipour, M.; Martinez-de-Hoyo, R. The efficacy and safety of Favipiravir in treatment of COVID-19: A systematic review and meta-analysis of clinical trials. Sci. Rep. 2021, 11, 11022.
- Maxwell, D.; Sanders, K.C.; Sabot, O.; Hachem, A.; Llanos-Cuentas, A.; Olotu, A.; Gosling, R.; Cutrell, J.B.; Hsiang, M.S. COVID-19 Therapeutics for Low- and Middle-Income Countries: A Review of Candidate Agents with Potential for Near-Term Use and Impact. Am. J. Trop. Med. Hyg. 2021, 105, 584–595.
- Salgado-Benvindo, C.; Thaler, M.; Tas, A.; Ogando, N.S.; Bredenbeek, P.J.; Ninaber, D.K.; Wang, Y.; Hiemstra, P.S.; Snijder, E.J.; van Hemert, M.J. Suramin Inhibits SARS-CoV-2 Infection in Cell Culture by Interfering with Early Steps of the Replication Cycle. Antimicrob. Agents Chemother. 2020, 64, e00900–e00920.
- Yin, W.; Luan, X.; Li, Z.; Zhou, Z.; Wang, Q.; Gao, M.; Wang, X.; Zhou, F.; Shi, J.; You, E.; et al. Structural basis for inhibition of the SARS-CoV-2 RNA polymerase by suramin. Nat. Struct. Mol. Biol. 2021, 28, 319–325.
- Venkataraman, S.; Prasad, B.; Selvarajan, R. RNA Dependent RNA Polymerases: Insights from Structure, Function and Evolution. Viruses 2018, 10, 76.
- Li, H.; Dong, J.; Cai, M.; Xu, Z.; Cheng, X.-D.; Qin, J.-J. Protein degradation technology: A strategic paradigm shift in drug discovery. J. Hematol. Oncol. 2021, 14, 138.
- Costales, M.G.; Childs-Disney, J.L.; Haniff, H.S.; Disney, M.D.J. How we think about targeting RNA with small molecules. J. Med. Chem. 2020, 63, 8880–8900.
- Pettersson, M.; Crews, C.M. PROteolysis TArgeting Chimeras (PROTACs)—Past, present and future. Drug Discov. Today Technol. 2019, 31, 15–27.
- Smith, B.E.; Wang, S.L.; Jaime-Figueroa, S.; Harbin, A.; Wang, J.; Hamman, B.D.; Crews, C.M. Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat. Commun. 2019, 10, 131.
- Yang, Z.; Sun, Y.; Ni, Z.; Yang, C.; Tong, Y.; Liu, Y.; Li, H.; Rao, Y. Merging PROTAC and molecular glue for degrading BTK and GSPT1 proteins concurrently. Cell Res. 2021, 31, 1315–1318.
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200.
- Nganvongpanit, K.; Müller, H.; Rings, F. Targeted suppression of E-cadherin gene expression in bovine preimplantation embryo by RNA interference technology using double-stranded RNA. Mol Reprod Dev. 2006, 73, 153–163.
- Schluep, T.; Lickliter, J.; Hamilton, J.; Lewis, D.L.; Lai, C.L.; Lau, J.Y.; Locarnini, S.A.; Gish, R.G.; Given, B.D. Safety, Tolerability, and Pharmacokinetics of ARC-520 Injection, an RNA Interference-Based Therapeutic for the Treatment of Chronic Hepatitis B Virus Infection, in Healthy Volunteers. Clin. Pharmacol. Drug Dev. 2017, 6, 350–362.
- Yuen, M.F.; Schiefke, I.; Yoon, J.H.; Ahn, S.H.; Heo, J.; Kim, J.H.; Lik Yuen Chan, H.; Yoon, K.T.; Klinker, H.; Manns, M.J.H. RNA Interference Therapy With ARC-520 Results in Prolonged Hepatitis B Surface Antigen Response in Patients With Chronic Hepatitis B Infection. Hepatology 2020, 72, 19–31.
- Yuen, M.-F.; Wong, D.K.-H.; Schluep, T.; Lai, C.-L.; Ferrari, C.; Locarnini, S.; Lo, R.C.-L.; Gish, R.G.; Hamilton, J.; Wooddell, C.I. Long-term serological, virological and histological responses to RNA inhibition by ARC-520 in Chinese chronic hepatitis B patients on entecavir treatment. Gut 2022, 71, 789–797.
- Agarwal, K.; Gane, E.; Cheng, W.; Sievert, W.; Roberts, S.; Ahn, S.H.; Kim, Y.J.; Streinu-Cercel, A.; Denning, J.; Symonds, W.J.H. HBcrAg, HBV-RNA declines in a phase 2a study evaluating the multi-dose activity of ARB-1467 in HBeAg-positive and negative virally suppressed subjects with hepatitis B. J. Hepatol. 2017, 66, S688–S689.
- Martinez, M.G.; Villeret, F.; Testoni, B.; Zoulim, F. Can we cure hepatitis B virus with novel directrect Symonds. Liver Int. 2020, 40, 27–34.
- Vaillant, A. HBsAg, subviral particles, and their clearance in establishing a functional cure of chronic hepatitis B virus infection. ACS Infect. Dis. 2020, 7, 1351–1368.
- Diab, A.; Foca, A.; Zoulim, F.; Durantel, D.; Andrisani, O. The diverse functions of the hepatitis B core/capsid protein (HBc) in the viral life cycle: Implications for the development of HBc-targeting antivirals. Antivir. Res. 2018, 149, 211–220.
- Nassal, M. Hepatitis B viruses: Reverse transcription a different way. Virus Res. 2008, 134, 235–249.
- Tan, Z.; Pionek, K.; Unchwaniwala, N.; Maguire, M.L.; Loeb, D.D.; Zlotnick, A.J. The interface between hepatitis B virus capsid proteins affects self-assembly, pregenomic RNA packaging, and reverse transcription. J. Virol. 2015, 89, 3275–3284.
- Venkatakrishnan, B.; Katen, S.P.; Francis, S.; Chirapu, S.; Finn, M.; Zlotnick, A.J. Hepatitis B virus capsids have diverse structural responses to small-molecule ligands bound to the heteroaryldihydropyrimidine pocket. J. Virol. 2016, 90, 3994–4004.
- Mak, L.-Y.; Wong, D.K.-H.; Seto, W.-K.; Lai, C.-L.; Yuen, M.F. Hepatitis B core protein as a therapeutic target. Expert Opin. Ther. Targets 2017, 21, 1153–1159.
- Schlicksup, C. Hepatitis B Virus Capsid Protein as an Antiviral Target; Indiana University: Bloomington, IN, USA, 2020.
- Stray, S.J.; Zlotnick, A. BAY 41-4109 has multiple effects on Hepatitis B virus capsid assembly. J. Mol. Recognit. JMR 2006, 19, 542–548.
- Chen, W.; Liu, F.; Zhao, Q.; Ma, X.; Lu, D.; Li, H.; Zeng, Y.; Tong, X.; Zeng, L.; Liu, J. Discovery of phthalazinone derivatives as novel hepatitis B virus capsid inhibitors. J. Med. Chem. 2020, 63, 8134–8145.
More
Information
Subjects:
Medicine, Research & Experimental
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
862
Revisions:
2 times
(View History)
Update Date:
14 Nov 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No