 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Kerstin Voigt | + 3431 word(s) | 3431 | 2020-11-04 07:27:46 | | | |
| 2 | Lily Guo | Meta information modification | 3431 | 2020-12-09 05:02:59 | | | | |
| 3 | Lily Guo | Meta information modification | 3431 | 2020-12-09 11:36:07 | | |
Video Upload Options
Iron is a key transition metal required by most microorganisms and is prominently utilised in the transfer of electrons during metabolic reactions. The acquisition of iron is essential and becomes a crucial pathogenic event for opportunistic fungi. Iron is not readily available in the natural environment as it exists in its insoluble ferric form, i.e., in oxides and hydroxides. During infection, the host iron is bound to proteins such as transferrin, ferritin, and haemoglobin. As such, access to iron is one of the major hurdles that fungal pathogens must overcome in an immunocompromised host. Thus, these opportunistic fungi utilise three major iron acquisition systems to overcome this limiting factor for growth and proliferation. (Draft for definition)
1. Introduction
In biology, iron is an essential micronutrient for almost all eukaryotes and most prokaryotes[1] . Iron is the fourth most abundant trace element in the environment, but the bioavailability (Fe2+) is limited due to oxidation into the insoluble ferric hydroxides (Fe3+) by atmospheric oxygen[2] [2]. In this state, iron has a solubility of approximately 10−9 M at neutral pH[3] [3]. Nonetheless, the involvement of iron in numerous important metabolic processes and as enzyme cofactors is due to its capacity for electron exchange[4] [4]. This transition metal is required in DNA, RNA and amino acid synthesis, oxygen transport, cellular respiration (iron-sulphur cluster (Fe-S) containing ferredoxins, haem-containing cytochromes), enzymatic reactions such as Fe-S proteins, e.g., fumarase and aconitase of the tricarboxylic acid cycle (TCA cycle)[5][6][7] [5–7]. Although it is a key trace element, iron also presents a danger to biological systems. Iron (Fe2+) triggered Fenton reaction produces reactive oxygen species (ROS) such as superoxide (O2•−), hydrogen peroxide (H2O2), and hydroxyl radicals (OH•) (Equation 1)[8] [8]. Hydroxyl radicals produced during these reactions are deleterious and can damage cellular components such as DNA, proteins, and lipids[9] [9]. Due to the redox property of iron, it is imperative that organisms have tightly regulated homeostatic mechanisms to maintain enough intracellular iron while actively avoiding the detrimental effects of excess iron[10] [10].
Fe3 + O2• à Fe2 + O2
Fe2+ + H2O2 à Fe3+ + OH• + OH−
Net Reaction: O2•− + H2O2 à OH• + OH− + O2
In low iron environments, cells employ strict iron usage called the iron-sparing response, which allows small concentrations to be used in essential enzymatic processes[11] [11]. High-affinity acquisition systems are expressed under these conditions, which allows for the rapid and efficient uptake of iron [12][3,12]. Under high-iron conditions, these uptake systems are repressed, and excess iron is stored in intracellular compartments, e.g., vacuole or ferritin in mucoralean fungi[3][13][14] [3,13–15]. [15]
In the host, iron is kept extremely low (i.e., < 10−24 M for Fe3+ in serum), and other trace metals, are usually bound to proteins[16] [16]. During infection, iron is further restricted by numerous host mechanisms[17] [17]. These mechanisms function by actively chelating extracellular Fe3+ to high-affinity iron-binding proteins such as glycoproteins, transferrin, and lactoferrin, including intracellular sequestration by haemoglobin, ferritin, cytochromes, and the hepcidin axis, to name a few[2][18] [2,18]. These elegant pathways and mechanisms for controlling systemic iron concentrations are known as nutritional immunity, and its importance in the host immune response to infections has been thoroughly described[2][17][19] [2,17,19]. [19][20]
Invading fungal pathogens must overcome these limitations to access host iron and other key metals such as zinc, copper, manganese, and nickel to proliferate and cause disease. As such, healthy individuals are usually not susceptible as their immune system is robust[17] [17]. On the other hand, fungal pathogens can cause debilitating and devasting diseases to various patient groups, especially among those who are immunocompromised or hospitalised with severe underlying conditions[20][21] [20,21]. Those at high risk include patients undergoing haematopoietic stem cell (HSCT), solid organ transplant recipients (SOTs), AIDS patients, those receiving antilymphocyte monoclonal antibodies, and other immunomodulators, as well as patients with other underlying diseases associated with immune dysfunction[20][21] [20,21]. Opportunistic fungal infections are underappreciated in comparison to bacterial, viral, and parasitic infections[22] [22]. With the current advancements in medicine and the increasing cohort of immunosuppressed individuals, the mortality rate caused by fungal infections is on a constant rise[23] [23]. For instance, Candida albicans and other Candida species are the most common fungal pathogens responsible for superficial mucosal infections as well as life-threatening systemic diseases[24] [24]. Cryptococcus neoformans is the most important opportunistic pathogen in HIV/AIDS patients. Although access to antiretroviral therapy (ART) has improved globally, the number of cryptococcal infections remains high, with an estimated 278,000 reported cases worldwide and a mortality rate of approximately 81%[24][25][26][27][28] [24–28]. Aspergillus fumigatus and other pathogenic Aspergillus species cause a wide spectrum of diseases known as aspergilloses. These include allergic bronchopulmonary, chronic pulmonary, and invasive aspergillosis[29] [29]. As fungi cause serious opportunistic infections, there is a new precedent for novel approaches in treatment options, as the range remains limited and there are increasing reports of resistance[30] [30].
2. The Reductive System for Iron Uptake
The mechanism for iron acquisition and homeostasis has been well documented in the model organism Saccharomyces cerevisiae, which established the foundations for further studies in fungal pathogens[31] [34]. There are two main mechanisms for iron uptake in S. cerevisiae, the reductive high affinity (HA) and non-reductive systems[32][33] [35,36]. The reductive HA pathway involves three sequential steps: (i) the initial reduction of ferric (Fe3+) to ferrous (Fe2+) iron by a dedicated membrane-bound ferric reductase encoded by FRE1 and FRE2 genes; (ii) the re-oxidation to ferric iron (Fe3+) by the multicopper ferroxidase (ferroxidase) encoded by the FET3 gene; and (iii) the import of the insoluble ferric iron (Fe3+) by the high-affinity iron permease encoded by the FTR1 gene[34][35][36] [37–39] (Figure 1). The non-reductive system involves the use of siderophores (xenosiderophores) that bind iron, which are then translocated across the membrane via specific/specialised transporters. This will be discussed later [34][37][38][37,40,41].
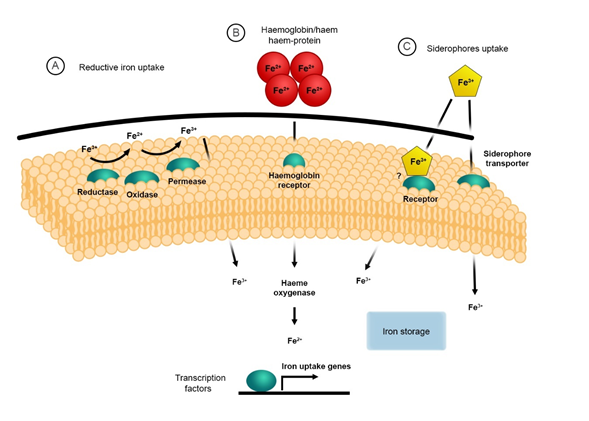
Fungal pathogens such as C. neoformans, C. albicans and A. fumigatus as well as pathogenic Mucorales, i.e., Rhizopus arrhizus (syn. R. oryzae, R. delemar), Mucor circinelloides and Lichtheimia corymbifera possess a reductive iron uptake system[39] [42]. This system has highly conserved orthologs of the three major components, i.e., surface ferric reductases, ferroxidases and permeases similar to those identified in S. cerevisiae [31][32][40][34,35,43]. For these pathogens, the reductive HA pathway is important for releasing ferric iron bound to other complexes, e.g., transferrin, ferritin, or siderophores[41][42][43][44][45]44–48]. The latter organism, L. corymbifera, has recently been shown to have conserved orthologs belonging to this system[46] [49]. It has been demonstrated that the ferric reductases are also involved in intracellular iron transport and storage of iron when present on the vacuole membranes[47][48] [50,51]. The ferric reductases encoded by the FRE genes are integral membrane proteins that require NADPH, flavin mononucleotides (FMN), and haem for their activity. The oxidation of cytoplasmic NADPH is catalysed by these ferric reductases, which then transfer the electron across the plasma membrane to facilitate the reduction of metals, e.g., iron[49][50][51] [52–54]. It has also been shown that these reductases have cupric reductase activity, and they can facilitate the use of siderophore-bound iron [32][41][42][49][52][35,44,45,52,55]. Eight putative ferric reductases have been identified in C. neoformans; these are FRE1–FRE7 and FRE201[49] [52]. The transcription of FRE2 and FRE4 is regulated by FeCl3 or haemin, which indicates that these genes may have an important role in iron homeostasis during iron-starvation. Saika et al., 2014 demonstrated that Fre2 is essential for fungal growth in the presence of transferrin and haem and contributed to virulence in mouse inhalation model of cryptococcosis[49] [52]. Copper also plays a role in the transcriptional regulation of the FRE genes in C. neoformans, C. albicans, and S. cerevisiae[49][50][53][54][55] [52,53,56–58].
As previously mentioned, the next stage in the reductive iron uptake system involves the transport of the reduced iron by the high-affinity ferric transporters. This transport system requires the dual-protein complex consisting of the ferroxidase Fet3 and the permease Ftr1. The ferroxidase, Fet3, catalyses the oxidation of ferrous (Fe2+) to ferric iron (Fe3+), which is immediately transported into the cell by the permease Ftr1[56] [59]. Components of the reductive iron uptake system have been identified and characterised in numerous opportunistic fungal pathogens, most of which are thoroughly summarised in the following review[57] [60]. Characterised and putative homologs of the reductive pathway components have been identified in pathogenic Mucorales and are summarised in Table 1. To date, five genes—FET3, FET31, FET33, FET34, and FET99—have been identified in C. albicans that are orthologs to the S. cerevisiae FET3 gene[41][58][44,61]. Under iron starvation, it has been shown that both FET3 and FET34 and the permease FTR1 are regulated. FET34 has an important role in iron acquisition, hyphal growth, and virulence in murine models of systemic candidiasis [59][62]. Ftr1 and the ferric reductase Fre10 may be involved in iron acquisition from host proteins, i.e., ferritin and transferrin [42][60][61][45,63,64]. Interestingly, virulence in a mouse model of systemic candidiasis is attenuated in FTR1 knockouts, and this strongly indicates that the permeases are key virulence determinants[3][61] [3,44,45,63].
Table 1. Reductive iron acquisition system in S. cerevisiae and pathogenic Mucoralean species.
|
Component |
Species |
Gene |
Functions |
Ref |
|
Ferric reductases |
S. cerevisiae |
FRE1, FRE2 |
Ferric iron reduction at the cell surface |
|
|
Rhizopus spp |
FRE (homolog) |
Putative protein—ferric iron reduction at the cell surface |
||
|
M. circinelloides |
FRE (homolog) |
Putative protein—ferric iron reduction at the cell surface |
||
|
L. corymbifera |
FRE5 (homolog)–three copies |
Putative protein—ferric iron reduction at the cell surface |
[46][49] |
|
|
Multicopper ferroxidase |
S. cerevisiae |
FET3 |
Multicopper-oxidase Ferrous iron oxidation and high-affinity uptake coupled with Ftr1 (permease) |
|
|
Rhizopus spp |
FET3 homolog |
Putative multicopper oxidase |
[45][48] |
|
|
M. circinelloides |
FETA, FETB, FETC |
Ferrous iron oxidation and high-affinity iron uptake |
[44][47] |
|
|
L. corymbifera |
FET3/5 homolog–three copies |
Putative multicopper oxidase |
[46][49] |
|
|
Iron permease |
S. cerevisiae |
FTR1 |
High-affinity iron uptake, coupled with FET3 (multicopper oxidase) |
|
|
Rhizopus spp |
FTR1 |
High affinity iron permease |
||
|
M. circinelloides |
FTR1 (homolog) |
Putative iron permease |
||
|
L. corymbifera |
FTR1 (homolog)—four copies |
Putative iron permease |
[46][49] |
The components of the reductive iron uptake system are also present in A. fumigatus. These include the cell-surface ferric reductases, ferroxidases (FetC), and the iron permease (FtrA). Like C. albicans Ftr1, the FTRA gene of A. fumigatus is also expressed under iron starvation. Mutants with an inactivated FTRA gene showed no difference in growth on iron-depleted medium and in virulence models compared to wild-type A. fumigatus, thereby indicating that the permease is not a virulence factor in A. fumigatus[71][72] [74,75]. In Mucorales, this system was shown to be strongly regulated, particularly in low iron conditions[44][45] [47,48]. Recently, it was demonstrated that there is overexpression of the ferroxidases (FET3) in the lung of mice confronted with invasive M. circinelloides [44][47]. In addition, there are three characterised copies of FET3 (Table 1.) in M. circinelloides, which were identified as FET3A, FET3B, and FET3C, with the latter being the most important for infection [47]. Single and double knockout strains of the FET3 genes were also shown to be critical components involved in iron uptake, particularly in low iron conditions both in vitro and in vivo[44] [47]. In R. delemar, the complete deletion of the iron permease (FTR1) results in reduced virulence[45][64] [48,66]. Interestingly, iron starvation induces the metacaspase dependent apoptotic response in strains lacking FTR1[45] [48]. In addition, there remains the possibility that the reductive pathway and the iron permeases (Ftr1) in Mucorales may also have a role in scavenging iron from other host proteins, e.g., ferritin or transferrin [64][64][40,64,76]. These examples highlight the importance of the reductive pathway has in survival and virulence under iron starvation.
Iron Acquisition and Susceptibility to Antifungals: implications in Therapy
Antifungal Treatment and Iron Chelation Therapy
Successful management of IFIs are based on the timely initiation of optimal antifungal therapy, reversal or discontinuation of underlying predisposing factors and the use of relevant adjunctive therapies[73] [222]. Additionally, immediate correction of metabolic disorders or abnormalities in patients with uncontrolled diabetes is mandatory in suspected mucormycosis cases. Surgical intervention for the complete removal of infected tissue in urgent cases significantly improves patient outcome[74][75][76][77] [199,202,204,223]. Only four classes of antifungal medications are currently available for the treatment of IFIs, these are: polyenes, pyrimidine analogue, echinocandins and triazoles[78][79][80] [207,210,224]. The latter i.e., echinocandins and azoles will be discussed later as emerging resistance is becoming more prevalent [81][225]. The first line treatment of invasive candidiasis is typically the echinocandins as well as formulations of amphotericin B (AMB)[82] [205]. For Cryptococcal infections, the gold standard antifungal drugs include the polyenes, flucytosine (5-FC), triazoles and their combinations [83][84][226,227]. Treatment options for invasive aspergillosis include voriconazole, liposomal amphotericin B (LAMB) and most recently isavuconazole[74][85][86] [199,228,229]. In mucormycosis, the lipid formulations of AMB, i.e., LAMB and AMB lipid complex, (AMLC) is the optimal treatment option[74][75][76][73][85][87][85] [199,202,204,222,228,230]. It is important to note that Mucoralean fungi are innately resistant to most antifungals in vitro, including voriconazole[88] [231]. Most recently, posaconazole and isavuconazole have exhibited activity against Mucorales [85][88][89][228,231,232]
Currently, therapeutic strategies to combat complicated infections as well as innate, emerging resistance in fungal pathogens include adjunctive therapies and new antifungal drugs[90] [233]. Adjunctive therapies functions by interfering with resistance mechanisms or modifying drug activity[91] [197]. Examples of the former include efflux pump inhibitors, which increase intracellular antifungal concentration, and histone deacetylase inhibitors, which are used in combination with azoles to inhibit fungal growth[91][92][93][94] [197,234–236]. Compounds that modify antifungal activity usually act synergistically by altering the fungal stress response mechanisms[95][96][97][98] [237–240]. These include statins, heat-shock protein 90 (Hsp90) inhibitors, nonsteroidal anti-inflammatory drugs, inhibitors of calcineurin and calmodulin, calcium homeostasis, selective serotonin reuptake, and iron homeostasis[91][78] [197,207].
Iron metabolism holds a central role in fungal pathogenesis, particularly in the development of mucormycosis. Thus, there is the possibility to use iron chelators as an adjunctive therapy strategy as this could limit/inhibit fungal growth. The iron chelator deferasirox is used for the treatment of iron overload in immunocompromised patients and those with elevated serum iron, e.g., diabetic & DKA patients. Preclinical data on DKA murine models of R. oryzae (R. arrhizus) infection found that treatment with deferasirox was as effective as LAMB therapy and combination treatment, i.e., deferasirox-LAMB, acted synergistically to improve survival[99][100][101][102][103] [31,101,143,241,242]. Although this showed promise, in the clinical application of deferasirox-LAMB, it was demonstrated to significantly increase mortality in patients with hematologic malignancies[100] [101,210,241,243,244]. On the other hand, this treatment strategy remains a viable option for other high-risk patient groups, e.g., DKA patients [200,202]. Deferasirox was also seen to enhance LAMP treatment in a murine model of invasive pulmonary aspergillosis. However, relevant clinical applications or data remain lacking [242,245]. Synergy was shown with fluconazole, ketoconazole, or AMB when combined with other iron chelators, including deferiprone, lactoferrin, and ciclopirox. These combinations proved successful in inhibiting A. fumigatus growth in vitro [237]. Another potential novel target for the treatment of Mucorales include the inhibition or blocking of the proteins involved in the reductive pathway. Antibodies targeting the iron permeases (Ftr1) of R. oryzae (R. arrhizus) protected DKA mice from infection [33,72,246]. Additionally, antibodies targeting the unique host proteins involved in receptor mediated endocytosis of fungal spores, i.e., 78kDa glucose-regulated protein (Grp78/HspA5) are possible targets. Grp78/HspA5 is overexpressed in patients with hyperglycaemia, DKA, and elevated serum iron; thus, antibodies, i.e. anti-Grp78 may be promising novel targets as it was shown to offer protection in a murine DKA model. Similar protective attributes were seen when antibodies of the fungal spore coat protein H or CotH i.e., anti-CotH (the interaction partner of Grp78/HspA5) were used in DKA murine model [105,231,247].
Antifungal Resistance and Iron
Echinocandins
Antifungal compounds that specifically target the cell wall components include Ibrexafungerp (SCY-078) and the Echinocandins, e.g., caspofungin, micafungin, and anidulafungin [181,248]. Ibrexafungerp (SCY-078) functions by actively inhibiting the 1,3-β-D-glucan synthase while the Echinocandins inhibit the 1,3-β-D-glucan synthase by noncompetitively binding to the Fksp subunit of the enzyme, which leads to a decrease in the amount of β-D-glucans present in the cell wall (Figure 5.) [249–254]. Cell death is seen in C. albicans when this enzyme is inhibited by caspofungin and micafungin [252,254–257]. Interestingly, ΔCCC2 cells (defectives in copper transport) show hypersensitivity to echinocandins [258]. On the other hand, elevated proportions of chitin in the cell wall of Candida species exhibit increased resistance to caspofungin, particularly in in vivo candidiasis models [238,248,251,259]. Recently, Pradhan et al. 2019 demonstrated that iron-limitations induces a β-glucan masking phenotype as well as cell wall remodelling and thickening. However, defects in this phenotype was observed in mutants lacking the permease and transcription factor (ΔFTR1 and ΔSEF1, respectively) [64]. Through this β-glucan masking, there is reduced phagocytosis and a dramatic reduction in proinflammatory cytokines (TNF-α and IL-6) produced by peripheral blood mononuclear cells (PBMCs) [64,163,164]. However, the use of caspofungin enhances β-glucan exposure [163,164]. Interestingly, the 1-3-β-d-Glucan inhibitor ibrexafungerp appears to be effective against clinical isolates that are resistant to echinocandins [260,261]. The dynamic nature of the cell wall has a major role in the development of antifungal resistance [262,263]. In both C. albicans and A. fumigatus, changes in the structural composition of the cell wall have been noted in strains showing antifungal resistance [210,213,214].
Azoles
Azole antifungals have been in clinical use for more than 20 years [264]. The azoles are separated into two distinctive classes, i.e., the triazoles and the imidazoles. Triazoles used in the clinical setting include fluconazole, itraconazole, voriconazole and posaconazole [265]. Common imidazoles used are clotrimazole, ketoconazole and miconazole [265,266]. Cytochrome P450 (CYP450) is an enzyme that converts lanosterol to ergosterol, which is the major sterol in the fungal plasma membrane. Azoles inhibit the CYP450 enzymes which causes increase permeability of the fungal plasma membrane (Figure 5.) [265–267]. Azoles also affect other efflux transporters, including major facilitator superfamily (MFS) transporters and ATP-binding cassette (ABC) transporters [268]. Susceptibility to azole antifungals is seen in Candida spp, C. neoformans, Aspergillus spp, and the Mucorales, to name a few. However, resistance has also been well characterised among this class of antifungal therapy [239,268–271]. The direct target of fluconazole is Erg11 (homologous to the yeast CYP51 F5), an enzyme involved in the ergosterol biosynthesis pathway [97,240,272,273].
In C. albicans, it was shown that intracellular iron depletion leads to increased fluidity of the plasma membrane as there is reduced ergosterol [240,274]. Gene expression of ERG11, which encodes for lanosterol 14-α demethylase as well as the ERG3 gene, which encodes for the Δ5,6-desaturase is affected by intracellular iron availability (Figure 5.). Erg3 catalyses the addition of a carbon-carbon double bond to the substrate molecules in the finals steps of the ergosterol biosynthesis pathway [39,240,271,275]. The strains lacking the high-affinity iron permease Ftr1 (ΔFTR1), null mutants (lacking both: ΔFTR1 and ΔFTR2) as well as ΔCCC2 mutants (copper transporter) were all shown to be more susceptible to fluconazole [240]. An important note is that the Ccc2 copper transporter is responsible for the copper acquisition, as copper is a key component of the multicopper oxidase (Fet3) protein in the reductive pathway [240,258]. Iron deprivation results in the downregulation of ERG11 [240,271]. As such, the increased membrane fluidity due to lower ergosterol content seen in the iron uptake mutants (ΔFTR1, ΔFTR2, ΔFTR1 ΔFTR2, and ΔCCC2) leads to higher passive diffusion of azole antifungals, thus increased susceptibility [271,276]. This is compounded by the upregulation of ERG3, which in an azole-inhibited pathway, allows for the accumulation of toxic intermediates [39,271]. Therefore, Erg3 acts synergistically with azoles increasing susceptibility [239,240,271]. On the other hand, mutations or deletions of the ERG3 gene, as well as upregulation of ERG11, confers azole resistance in C. albicans (Figure 5.) [271,275,277]. Similarly, the reductive iron uptake system in C. neoformans has an important role in resistance to azoles [97,138,278]. Mutants lacking both the multicopper ferroxidase (CFO1) and the iron permease (CFT1) had reduced intracellular iron levels, which significantly increase azole drug susceptibility, i.e., to fluconazole [97,279]. Interestingly, overexpression of ERG11 in CFO1 mutants exhibited reduced susceptibility to fluconazole [97,272,277,280]. Innate and acquired reduced susceptibility and resistance to azole in A. fumigatus has been linked to numerous point mutations in the CYP51A gene [268,270,281]. It has been demonstrated that the Mucorales have an intrinsic resistance to azole antifungals, specifically to the short-tailed azoles, i.e., fluconazole and voriconazole [270,281–284]. It was found that this intrinsic resistance may be caused by an amino acid substitution in the cytochrome P51 or CYP51 F5 (Erg11) enzyme; changing a Tryosine (Y) to Phenylalanine (F) at position 129 i.e., Y129F [270]. Interestingly, the CYP51 enzyme was shown to be highly regulated by iron in A. fumigatus [268,279].
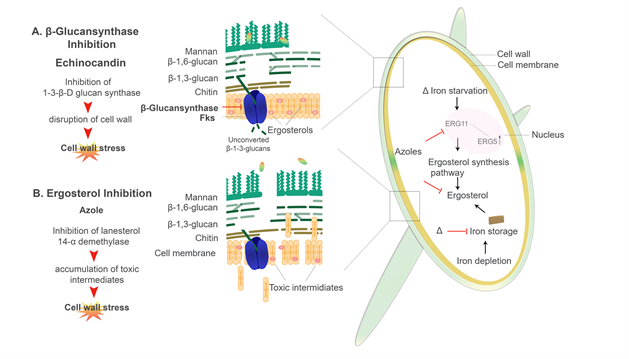
Figure 5. Azoles and echinocandin antifungal drugs and their mechanism of actions: An illustration of two main classes of antifungal drugs used clinically and how they affect the fungal cell of C. albicans. (A) Echinocandins, e.g., caspofungin, inhibit β-(1-3)-D-glucan synthase in the cell membrane, which leads to disruption in cell wall integrity. (B) Azoles, e.g., fluconazole, inhibit Erg11/CYP51 F5, which blocks the production of ergosterol, leading to the accumulation of toxic sterol intermediates. Δ indicates where iron starvation or depletion may contribute to increased susceptibility to azole antifungals.
References
- Domenico: I. De; Ward, D.M.; Kaplan, J. Regulation of iron acquisition and storage : consequences for iron-linked disorders. 2008, 9, 72–81, doi:10.1038/nrm2295.
- Cassat, J.E.; Skaar, E.P. Iron in Infection and Immunity. Cell Host Microbe 2013, 13, 509–519, doi:10.1016/j.chom.2013.04.010.
- Philpott, C.C. Iron uptake in fungi: A system for every source. Biochim. Biophys. Acta - Mol. Cell Res. 2006, 1763, 636–645, doi:10.1016/j.bbamcr.2006.05.008.
- Boyce, K.J.; Andrianopoulos, A. Fungal dimorphism: The switch from hyphae to yeast is a specialized morphogenetic adaptation allowing colonization of a host. FEMS Microbiol. Rev. 2015, 39, 797–811, doi:10.1093/femsre/fuv035.
- Beard, J.L. Iron Biology in Immune Function, Muscle Metabolism and Neuronal Functioning. J. Nutr. 2001, 131, 568S-580S, doi:10.1093/jn/131.2.568S.
- Krewulak, K.D.; Vogel, H.J. Structural biology of bacterial iron uptake. Biochim. Biophys. Acta - Biomembr. 2008, 1778, 1781–1804, doi:10.1016/j.bbamem.2007.07.026.
- Rouault, T.A.; Tong, W.H. Iron–sulfur cluster biogenesis and human disease. Trends Genet. 2008, 24, 398–407, doi:10.1016/j.tig.2008.05.008.
- Prousek, J. Fenton chemistry in biology and medicine. In Proceedings of the Pure and Applied Chemistry; 2007; Vol. 79, pp. 2325–2338.
- Halliwell, B.; Gutteridget, J.M.C. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem. J. 1984, 219, 1–14.
- Brault, A.; Mourer, T.; Labbé, S. Molecular basis of the regulation of iron homeostasis in fission and filamentous yeasts. IUBMB Life 2015, 67, 801–815, doi:10.1002/iub.1441.
- Kaplan, J.; McVey Ward, D.; Crisp, R.J.; Philpott, C.C. Iron-dependent metabolic remodeling in S. cerevisiae. Biochim. Biophys. Acta - Mol. Cell Res. 2006, 1763, 646–651, doi:10.1016/j.bbamcr.2006.03.008.
- Miethke, M. Molecular strategies of microbial iron assimilation: from high-affinity complexes to cofactor assembly systems. Metallomics 2013, 5, 15–28, doi:10.1039/C2MT20193C.
- Ibrahim, A.S.; Spellberg, B.; Walsh, T.J.; Kontoyiannis, D.P. Pathogenesis of mucormycosis. Clin. Infect. Dis. 2012, 54, 1–7, doi:10.1093/cid/cir865.
- Comensoli, L.; Bindschedler, S.; Junier, P.; Joseph, E. Iron and Fungal Physiology: A Review of Biotechnological Opportunities. Adv. Appl. Microbiol. 2017, 98, 31–60, doi:10.1016/bs.aambs.2016.11.001.
- Carrano, C.J.; Böhnke, R.; Matzanke, B.F. Fungal ferritins: The ferritin from mycelia of Absidia spinosa is a bacterioferritin. FEBS Lett. 1996, 390, 261–264, doi:10.1016/0014-5793(96)00667-9.
- Doguer, C.; Ha, J.-H.; Collins, J.F. Intersection of Iron and Copper Metabolism in the Mammalian Intestine and Liver. In Comprehensive Physiology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2018; Vol. 176, pp. 1433–1461 ISBN 2163684814.
- Kehl-Fie, T.E.; Skaar, E.P. Nutritional immunity beyond iron: a role for manganese and zinc. Curr. Opin. Chem. Biol. 2010, 14, 218–224, doi:10.1016/j.cbpa.2009.11.008.
- Lane, D.J.R.; Merlot, A.M.; Huang, M.L.H.; Bae, D.H.; Jansson, P.J.; Sahni, S.; Kalinowski, D.S.; Richardson, D.R. Cellular iron uptake, trafficking and metabolism: Key molecules and mechanisms and their roles in disease. Biochim. Biophys. Acta - Mol. Cell Res. 2015, 1853, 1130–1144, doi:10.1016/j.bbamcr.2015.01.021.
- Hood, M.I.; Skaar, E.P. Nutritional immunity: transition metals at the pathogen–host interface. Nat. Rev. Microbiol. 2012, 10, 525–537, doi:10.1038/nrmicro2836.
- Pfaller, M. a; Diekema, D.J. Epidemiology of invasive mycoses in North America. Crit. Rev. Microbiol. 2010, 36, 1–53, doi:0.1128/JCM.42.10.4419–4431.2004.
- Sam, Q.H.; Yew, W.S.; Seneviratne, C.J.; Chang, M.W.; Chai, L.Y.A. Immunomodulation as Therapy for Fungal Infection: Are We Closer? Front. Microbiol. 2018, 9, 1–16, doi:10.3389/fmicb.2018.01612.
- Rodrigues, M.L.; Nosanchuk, J.D. Fungal diseases as neglected pathogens: A wake-up call to public health officials. PLoS Negl. Trop. Dis. 2020, 14, 1–9, doi:10.1371/journal.pntd.0007964.
- Low, C.Y.; Rotstein, C. Emerging fungal infections in immunocompromised patients. F1000 Med. Rep. 2011, 3, 1–8, doi:10.3410/M3-14.
- Jabra-Rizk, M.A.; Kong, E.F.; Tsui, C.; Nguyen, M.H.; Clancy, C.J.; Fidel, P.L.; Noverr, M. Candida albicans Pathogenesis: Fitting within the Host-Microbe Damage Response Framework. Infect. Immun. 2016, 84, 2724–2739, doi:10.1128/IAI.00469-16.
- Armstrong-James, D.; Meintjes, G.; Brown, G.D. A neglected epidemic: fungal infections in HIV/AIDS. Trends Microbiol. 2014, 22, 120–127, doi:10.1016/j.tim.2014.01.001.
- Warkentien, T.; Crum-Cianflone, N.F. An update on Cryptococcus among HIV-infected patients. Int. J. STD AIDS 2010, 21, 679–684, doi:10.1258/ijsa.2010.010182.
- Rajasingham, R.; Smith, R.M.; Park, B.J.; Jarvis, J.N.; Govender, N.P.; Chiller, T.M.; Denning, D.W.; Loyse, A.; Boulware, D.R. Global burden of disease of HIV-associated cryptococcal meningitis: an updated analysis. Lancet Infect. Dis. 2017, 17, 873–881, doi:10.1016/S1473-3099(17)30243-8.
- Franco-Paredes, C.; Chastain, D.B.; Rodriguez-Morales, A.J.; Marcos, L.A. Cryptococcal meningoencephalitis in HIV/AIDS: when to start antiretroviral therapy? Ann. Clin. Microbiol. Antimicrob. 2017, 16, 9, doi:10.1186/s12941-017-0184-2.
- Haas, H. Fungal siderophore metabolism with a focus on Aspergillus fumigatus. Nat. Prod. Rep. 2014, 31, 1266–1276, doi:10.1039/c4np00071d.
- Wiederhold, N.P. Antifungal resistance: current trends and future strategies to combat. Infect. Drug Resist. 2017, 10, 249–259, doi:10.2147/IDR.S124918.
- Haas, H. Iron - a key nexus in the virulence of Aspergillus fumigatus. Front. Microbiol. 2012, 3, 1–10, doi:10.3389/fmicb.2012.00028.
- Yun, C.; Ferea, T.; Rashford, J.; Ardon, O.; Brown, P.O.; Botstein, D.; Kaplan, J.; Philpott, C.C. Desferrioxamine-mediated Iron Uptake in Saccharomyces cerevisiae. J. Biol. Chem. 2000, 275, 10709–10715.
- Baker Brachmann, C.; Davies, A.; Cost, G.J.; Caputo, E.; Li, J.; Hieter, P.; Boeke, J.D. Designer deletion strains derived from Saccharomyces cerevisiae S288C: A useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 1998, 14, 115–132, doi:10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2.
- Hassett, R.F.; Romeo, A.M.; Kosman, D.J. Regulation of High Affinity Iron Uptake in the Yeast Saccharomyces cerevisiae. 1998, 273, 7628–7636, doi:10.1074/jbc.273.13.7628.
- Leal, S.M.; Roy, S.; Vareechon, C.; Carrion, S. deJesus J.; Clark, H.; Lopez-Berges, M.S.; DiPietro, A.; Schrettl, M.; Beckmann, N.; Redl, B.; et al. Targeting Iron Acquisition Blocks Infection with the Fungal Pathogens Aspergillus fumigatus and Fusarium oxysporum. PLoS Pathog. 2013, 9, e1003436, doi:10.1371/journal.ppat.1003436.
- Lindahl, P.A. A comprehensive mechanistic model of iron metabolism in: Saccharomyces cerevisiae. Metallomics 2019, 11, 1779–1799, doi:10.1039/c9mt00199a.
- Almeida, R.S.; Wilson, D.; Hube, B. Candida albicans iron acquisition within the host. FEMS Yeast Res. 2009, 9, 1000–1012, doi:10.1111/j.1567-1364.2009.00570.x.
- Gaensly, F.; Picheth, G.; Brand, D.; Bonfim, T.M.B. The uptake of different iron salts by the yeast Saccharomyces cerevisiae. Brazilian J. Microbiol. 2014, 45, 491–494, doi:10.1590/S1517-83822014000200016.
- Walther, G.; Wagner, L.; Kurzai, O. Updates on the taxonomy of mucorales with an emphasis on clinically important taxa. J. Fungi 2019, 5, doi:10.3390/jof5040106.
- Gerwien, F.; Skrahina, V.; Kasper, L.; Hube, B.; Brunke, S. Metals in fungal virulence. FEMS Microbiol. Rev. 2018, 42, 1–21, doi:10.1093/femsre/fux050.
- Knight, S.A.B.; Lesuisse, E.; Stearman, R.; Klausner, R.D.; Dancis, A. Reductive iron uptake by Candida albicans: Role of copper, iron and the TUP1 regulator. Microbiology 2002, 148, 29–40, doi:10.1099/00221287-148-1-29.
- Knight, S.A.B.; Vilaire, G.; Lesuisse, E.; Dancis, A. Iron Acquisition from Transferrin by Candida albicans Depends on the Reductive Pathway. Infect. Immun. 2005, 73, 5482–5492, doi:10.1128/IAI.73.9.5482-5492.2005.
- Blatzer, M.; Binder, U.; Haas, H. The metalloreductase FreB is involved in adaptation of Aspergillus fumigatus to iron starvation. Fungal Genet. Biol. 2011, 48, 1027–1033, doi:10.1016/j.fgb.2011.07.009.
- Navarro-Mendoza, M.I.; Pérez-Arques, C.; Murcia, L.; Martínez-García, P.; Lax, C.; Sanchis, M.; Capilla, J.; Nicolás, F.E.; Garre, V. Components of a new gene family of ferroxidases involved in virulence are functionally specialized in fungal dimorphism. Sci. Rep. 2018, 8, 1–13, doi:10.1038/s41598-018-26051-x.
- Andrianaki, A.M.; Kyrmizi, I.; Thanopoulou, K.; Baldin, C.; Drakos, E.; Soliman, S.S.M.; Shetty, A.C.; McCracken, C.; Akoumianaki, T.; Stylianou, K.; et al. Iron restriction inside macrophages regulates pulmonary host defense against Rhizopus species. Nat. Commun. 2018, 9, 3333, doi:10.1038/s41467-018-05820-2.
- Schwartze, V.U.; Winter, S.; Shelest, E.; Marcet-Houben, M.; Horn, F.; Wehner, S.; Linde, J.; Valiante, V.; Sammeth, M.; Riege, K.; et al. Gene Expansion Shapes Genome Architecture in the Human Pathogen Lichtheimia corymbifera: An Evolutionary Genomics Analysis in the Ancient Terrestrial Mucorales (Mucoromycotina). PLoS Genet. 2014, 10, doi:10.1371/journal.pgen.1004496.
- Urbanowski, J.L.; Piper, R.C. The iron transporter Fth1p forms a complex with the Fet5 iron oxidase and resides on the vacuolar membrane. J. Biol. Chem. 1999, 274, 38061–38070, doi:10.1074/jbc.274.53.38061.
- Haas, H.; Petrik, M.; Decristoforo, C. An Iron-Mimicking, Trojan Horse-Entering Fungi—Has the Time Come for Molecular Imaging of Fungal Infections? PLoS Pathog. 2015, 11, 1–7, doi:10.1371/journal.ppat.1004568.
- Saikia, S.; Oliveira, D.; Hu, G.; Kronstad, J. Role of ferric reductases in iron acquisition and virulence in the fungal pathogen Cryptococcus neoformans. Infect. Immun. 2014, 82, 839–850, doi:10.1128/IAI.01357-13.
- Georgatsou, E.; Mavrogiannis, L.A.; Fragiadakis, G.S.; Alexandraki, D. The yeast Fre1p/Fre2p cupric reductases facilitate copper uptake and are regulated by the copper-modulated Mac1p activator. J. Biol. Chem. 1997, 272, 13786–13792, doi:10.1074/jbc.272.21.13786.
- Georgatsou, E.; Alexandraki, D. Regulated expression of the Saccharomyces cerevisiae Fre1p/Fre2p Fe/Cu reductase related genes. Yeast 1999, 15, 573–584, doi:10.1002/(SICI)1097-0061(199905)15:7<573::AID-YEA404>3.0.CO;2-7.
- Philpott, C.C.; Protchenko, O. Response to iron deprivation in Saccharomyces cerevisiae. Eukaryot. Cell 2008, 7, 20–27, doi:10.1128/EC.00354-07.
- Sun, T.S.; Ju, X.; Gao, H.L.; Wang, T.; Thiele, D.J.; Li, J.Y.; Wang, Z.Y.; Ding, C. Reciprocal functions of Cryptococcus neoformans copper homeostasis machinery during pulmonary infection and meningoencephalitis. Nat. Commun. 2014, 5, doi:10.1038/ncomms6550.
- Waterman, S.R.; Hacham, M.; Hu, G.; Zhu, X.; Park, Y.D.; Shin, S.; Panepinto, J.; Valyi-Nagy, T.; Beam, C.; Husain, S.; et al. Role of a CUF1/CTR4 copper regulatory axis in the virulence of Cryptococcus neoformans. J. Clin. Invest. 2007, 117, 794–802, doi:10.1172/JCI30006.
- Yamaguchi-Iwai, Y.; Serpe, M.; Haile, D.; Yang, W.; Kosman, D.J.; Klausner, R.D.; Dancis, A. Homeostatic regulation of copper uptake in yeast via direct binding of MAC1 protein to upstream regulatory sequences of FRE1 and CTR1. J. Biol. Chem. 1997, 272, 17711–17718, doi:10.1074/jbc.272.28.17711.
- Kwok, E.Y.; Severance, S.; Kosman, D.J. Evidence for iron channeling in the Fet3p-Ftr1p high-affinity iron uptake complex in the yeast plasma membrane. Biochemistry 2006, 45, 6317–6327, doi:10.1021/bi052173c.
- Bairwa, G.; Hee Jung, W.; Kronstad, J.W. Iron acquisition in fungal pathogens of humans. Metallomics 2017, 9, 215–227, doi:10.1039/c6mt00301j.
- Protchenko, O.; Ferea, T.; Rashford, J.; Tiedeman, J.; Brown, P.O.; Botstein, D.; Philpott, C.C. Three Cell Wall Mannoproteins Facilitate the Uptake of Iron in Saccharomyces cerevisiae. J. Biol. Chem. 2001, 276, 49244–49250, doi:10.1074/jbc.M109220200.
- Cheng, X.; Xu, N.; Yu, Q.; Ding, X.; Qian, K.; Zhao, Q.; Wang, Y.; Zhang, B.; Xing, L.; Li, M. Novel insight into the expression and function of the multicopper oxidases in Candida albicans. Microbiology 2013, 159, 1044–1055, doi:10.1099/mic.0.065268-0.
- Ramanan, N.; Wang, Y. A high-affinity iron permease essential for Candida albicans virulence. Science 2000, 288, 1062–4, doi:10.1126/science.288.5468.1062.
- Pradhan, A.; Avelar, G.M.; Bain, J.M.; Childers, D.; Pelletier, C.; Larcombe, D.E.; Shekhova, E.; Netea, M.G.; Brown, G.D.; Erwig, L.; et al. Non-canonical signalling mediates changes in fungal cell wall PAMPs that drive immune evasion. Nat. Commun. 2019, 10, 1–14, doi:10.1038/s41467-019-13298-9.
- Yun, C.W.; Bauler, M.; Moore, R.E.; Klebba, P.E.; Philpott, C.C. The Role of the FRE Family of Plasma Membrane Reductases in the Uptake of Siderophore-Iron in Saccharomyces cerevisiae. J. Biol. Chem. 2001, 276, 10218–10223, doi:10.1074/jbc.M010065200.
- Severance, S.; Chakraborty, S.; Kosman, D.J. The Ftr1p iron permease in the yeast plasma membrane: Orientation, topology and structure-function relationships. Biochem. J. 2004, 380, 487–496, doi:10.1042/BJ20031921.
- Fu, Y.; Lee, H.; Collins, M.; Tsai, H.F.; Spellberg, B.; Edwards, J.E.; Kwon-Chung, K.J.; Ibrahim, A.S. Cloning and functional characterization of the Rhizopus oryzae high affinity iron permease (rFTR1) gene. FEMS Microbiol. Lett. 2004, 235, 169–176, doi:10.1016/j.femsle.2004.04.031.
- Trieu, T.A.; Navarro-Mendoza, M.I.; Pérez-Arques, C.; Sanchis, M.; Capilla, J.; Navarro-Rodriguez, P.; Lopez-Fernandez, L.; Torres-Martínez, S.; Garre, V.; Ruiz-Vázquez, R.M.; et al. RNAi-Based Functional Genomics Identifies New Virulence Determinants in Mucormycosis. PLOS Pathog. 2017, 13, e1006150, doi:10.1371/journal.ppat.1006150.
- Singh, A.; Severance, S.; Kaur, N.; Wiltsie, W.; Kosman, D.J. Assembly, activation, and trafficking of the Fet3p•Ftr1p high affinity iron permease complex in Saccharomyces cerevisiae. J. Biol. Chem. 2006, 281, 13355–13364, doi:10.1074/jbc.M512042200.
- Stearman, R.; Yuan, D.S.; Yamaguchi-Iwai, Y.; Klausner, R.D.; Dancis, A. A Permease-Oxidase Complex Involved in High-Affinity Iron Uptake in Yeast. Science (80-. ). 1996, 271, 1552–1557, doi:10.1126/science.271.5255.1552.
- Liu, M.; Lin, L.; Gebremariam, T.; Luo, G.; Skory, C.D.; French, S.W.; Chou, T.-F.; Edwards, J.E.; Ibrahim, A.S. Fob1 and Fob2 Proteins Are Virulence Determinants of Rhizopus oryzae via Facilitating Iron Uptake from Ferrioxamine. PLOS Pathog. 2015, 11, e1004842, doi:10.1371/journal.ppat.1004842.
- Ibrahim, A.S.; Gebremariam, T.; Lin, L.; Luo, G.; Husseiny, M.I.; Skory, C.D.; Fu, Y.; French, S.W.; Edwards, J.E.; Spellberg, B. The high affinity iron permease is a key virulence factor required for Rhizopus oryzae pathogenesis. Mol. Microbiol. 2010, 77, 587–604, doi:10.1111/j.1365-2958.2010.07234.x.
- López-Fernández, L.; Sanchis, M.; Navarro-Rodríguez, P.; Nicolás, F.E.; Silva-Franco, F.; Guarro, J.; Garre, V.; Navarro-Mendoza, M.I.; Pérez-Arques, C.; Capilla, J. Understanding Mucor circinelloides pathogenesis by comparative genomics and phenotypical studies. Virulence 2018, 9, 707–720, doi:10.1080/21505594.2018.1435249.
- Schrettl, M.; Bignell, E.; Kragl, C.; Joechl, C.; Rogers, T.; Arst, H.N.; Haynes, K.; Haas, H. Siderophore Biosynthesis But Not Reductive Iron Assimilation Is Essential for Aspergillus fumigatus Virulence. J. Exp. Med. 2004, 200, 1213–1219, doi:10.1084/jem.20041242.
- Schrettl, M.; Bignell, E.; Kragl, C.; Sabiha, Y.; Loss, O.; Eisendle, M.; Wallner, A.; Arst, H.N.; Haynes, K.; Haas, H. Distinct roles for intra- and extracellular siderophores during Aspergillus fumigatus infection. PLoS Pathog. 2007, 3, 1195–1207, doi:10.1371/journal.ppat.0030128.
- Tissot, F.; Agrawal, S.; Pagano, L.; Petrikkos, G.; Groll, A.H.; Skiada, A.; Lass-Flörl, C.; Calandra, T.; Viscoli, C.; Herbrecht, R. ECIL-6 guidelines for the treatment of invasive candidiasis, aspergillosis and mucormycosis in leukemia and hematopoietic stem cell transplant patients. Haematologica 2017, 102, 433–444, doi:10.3324/haematol.2016.152900.
- Carlesse, F.; Daudt, L.E.; Seber, A.; Dutra, Á.P.; Melo, A.S. de A.; Simões, B.; Macedo, C.R.D.; Bonfim, C.; Benites, E.; Gregianin, L.; et al. A consensus document for the clinical management of invasive fungal diseases in pediatric patients with hematologic cancer and/or undergoing hematopoietic stem cell transplantation in Brazilian medical centers. Brazilian J. Infect. Dis. 2019, 23, 395–409, doi:10.1016/j.bjid.2019.09.005.
- Sipsas, N. V.; Gamaletsou, M.N.; Anastasopoulou, A.; Kontoyiannis, D.P. Therapy of mucormycosis. J. Fungi 2018, 4, 1–17, doi:10.3390/jof4030090
- Cornely, O.A.; Arikan-Akdagli, S.; Dannaoui, E.; Groll, A.H.; Lagrou, K.; Chakrabarti, A.; Lanternier, F.; Pagano, L.; Skiada, A.; Akova, M.; et al. ESCMID and ECMM joint clinical guidelines for the diagnosis and management of mucormycosis 2013. Clin. Microbiol. Infect. 2014, 20, 5–26, doi:10.1111/1469-0691.12371.
- Petrikkos, G.; Skiada, A.; Lortholary, O.; Roilides, E.; Walsh, T.J.; Kontoyiannis, D.P. Epidemiology and clinical manifestations of mucormycosis. Clin. Infect. Dis. 2012, 54, 23–34, doi:10.1093/cid/cir866.
- Sanguinetti, M.; Posteraro, B.; Beigelman-Aubry, C.; Lamoth, F.; Dunet, V.; Slavin, M.; Richardson, M.D. Diagnosis and treatment of invasive fungal infections: Looking ahead. J. Antimicrob. Chemother. 2019, 74, II27–II37, doi:10.1093/jac/dkz041.
- Skiada, A.; Lass-Floerl, C.; Klimko, N.; Ibrahim, A.; Roilides, E.; Petrikkos, G. Challenges in the diagnosis and treatment of mucormycosis. Med. Mycol. 2018, 56, S93–S101, doi:10.1093/mmy/myx101.
- Alastruey-Izquierdo, A.; Cuesta, I.; Walther, G.; Cuenca-Estrella, M.; Rodriguez-Tudela, J.L. Antifungal susceptibility profile of human-pathogenic species of Lichtheimia. Antimicrob. Agents Chemother. 2010, 54, 3058–3060, doi:10.1128/AAC.01270-09.
- Marquez, L.; Quave, C.L. Prevalence and Therapeutic Challenges of Fungal Drug Resistance: Role for Plants in Drug Discovery. Antibiotics 2020, 9, 150, doi:10.3390/antibiotics9040150.
- Colombo, A.L.; de Almeida Júnior, J.N.; Slavin, M.A.; Chen, S.C.A.; Sorrell, T.C. Candida and invasive mould diseases in non-neutropenic critically ill patients and patients with haematological cancer. Lancet Infect. Dis. 2017, 17, e344–e356, doi:10.1016/S1473-3099(17)30304-3.
- Perfect, J.R.; Dismukes, W.E.; Dromer, F.; Goldman, D.L.; Graybill, J.R.; Hamill, R.J.; Harrison, T.S.; Larsen, R.A.; Lortholary, O.; Nguyen, M.-H.; et al. Clinical Practice Guidelines for the Management of Cryptococcal Disease: 2010 Update by the Infectious Diseases Society of America. 2010, 291, doi:10.1086/649858.
- Mourad, A.; Perfect, J. Present and Future Therapy of Cryptococcus Infections. J. Fungi 2018, 4, 79, doi:10.3390/jof4030079.
- Marty, F.M.; Ostrosky-Zeichner, L.; Cornely, O.A.; Mullane, K.M.; Perfect, J.R.; Thompson, G.R.; Alangaden, G.J.; Brown, J.M.; Fredricks, D.N.; Heinz, W.J.; et al. Isavuconazole treatment for mucormycosis: A single-arm open-label trial and case-control analysis. Lancet Infect. Dis. 2016, 16, 828–837, doi:10.1016/S1473-3099(16)00071-2.
- Walsh, T.J.; Lutsar, I.; Driscoll, T.; Dupont, B.; Roden, M.; Ghahramani, P.; Hodges, M.; Groll, A.H.; Perfect, J.R. Voriconazole in the treatment of Aspergillosis, Scedosporiosis and other invasive fungal infections in children. Pediatr. Infect. Dis. J. 2002, 21, 240–248, doi:10.1097/00006454-200203000-00015.
- Shoham, S.; Magill, S.S.; Merz, W.G.; Gonzalez, C.; Seibel#, N.; Buchanan$, W.L.; Knudsen$, T.A.; Sarkisova$, T.A.; Walsh$, T.J. Primary treatment of zygomycosis with liposomal amphotericin B: analysis of 28 cases. Med. Mycol. 2010, 48, 511–517, doi:10.3109/13693780903311944.
- Lax, C.; Pérez-Arques, C.; Navarro-Mendoza, M.I.; Cánovas-Márquez, J.T.; Tahiri, G.; Pérez-Ruiz, J.A.; Osorio-Concepción, M.; Murcia-Flores, L.; Navarro, E.; Garre, V.; et al. Genes, Pathways, and Mechanisms Involved in the Virulence of Mucorales. Genes (Basel). 2020, 11, 317, doi:10.3390/genes11030317.
- Perkhofer, S.; Lechner, V.; Lass-Flörl, C. In Vitro Activity of Isavuconazole against Aspergillus Species and Zygomycetes According to the Methodology of the European Committee on Antimicrobial Susceptibility Testing. Antimicrob. Agents Chemother. 2009, 53, 1645–1647, doi:10.1128/AAC.01530-08.
- Pagano, L.; Caira, M.; Valentini, C.G.; Posteraro, B.; Fianchi, L. Current therapeutic approaches to fungal infections in immunocompromised hematological patients. Blood Rev. 2010, 24, 51–61, doi:10.1016/j.blre.2009.11.003.
- Formanek, P.E.; Dilling, D.F. Advances in the Diagnosis and Management of Invasive Fungal Disease. Chest 2019, 156, 834–842, doi:10.1016/j.chest.2019.06.032.
- Drew, R. Potential role of aerosolized amphotericin B formulations in the prevention and adjunctive treatment of invasive fungal infections. Int. J. Antimicrob. Agents 2006, 27, 36–44, doi:10.1016/j.ijantimicag.2006.03.018.
- Butts, A.; Palmer, G.E.; David Rogers, P. Antifungal adjuvants: Preserving and extending the antifungal arsenal. 2016, doi:10.1080/21505594.2016.1216283.
- Holmes, A.R.; Cardno, T.S.; Strouse, J.J.; Ivnitski-Steele, I.; Keniya, M. V; Lackovic, K.; Monk, B.C.; Sklar, L.A.; Cannon, R.D. Targeting efflux pumps to overcome antifungal drug resistance. Futur. Med. Chem 2016, 8, 1485–1501, doi:10.4155/fmc-2016-0050.
- Zarember, K.A.; Cruz, A.R.; Huang, C.-Y.; Gallin, J.I. Antifungal Activities of Natural and Synthetic Iron Chelators Alone and in Combination with Azole and Polyene Antibiotics against Aspergillus fumigatus. Antimicrob. Agents Chemother. 2009, 53, 2654–2656, doi:10.1128/AAC.01547-08.
- Walker, L.A.; Gow, N.A.R.; Munro, C.A. Elevated chitin content reduces the susceptibility of Candida species to caspofungin. Antimicrob. Agents Chemother. 2013, 57, 146–154, doi:10.1128/AAC.01486-12.
- Mansfield, B.E.; Oltean, H.N.; Oliver, B.G.; Hoot, S.J.; Leyde, S.E.; Hedstrom, L.; White, T.C. Azole drugs are imported by facilitated diffusion in Candida albicans and other pathogenic fungi. PLoS Pathog. 2010, 6, 1–11, doi:10.1371/journal.ppat.1001126.
- Prasad, T.; Chandra, A.; Mukhopadhyay, C.K.; Prasad, R. Unexpected Link between Iron and Drug Resistance of Candida spp.: Iron Depletion Enhances Membrane Fluidity and Drug Diffusion, Leading to Drug-Susceptible Cells. Antimicrob. Agents Chemother. 2006, 50, 3597–3606, doi:10.1128/AAC.00653-06.
- Ibrahim, A.S.; Spellberg, B.; Edwards, J. Iron acquisition: a novel perspective on mucormycosis pathogenesis and treatment. Curr. Opin. Infect. Dis. 2008, 21, 620–625, doi:10.1097/QCO.0b013e3283165fd1.
- Spellberg, B.; Walsh, T.J.; Kontoyiannis, D.P.; Edwards, Jr., J.; Ibrahim, A.S. Recent Advances in the Management of Mucormycosis: From Bench to Bedside. Clin. Infect. Dis. 2009, 48, 1743–1751, doi:10.1086/599105.
- Boelaert, J.R.; Van Cutsem, J.; De Locht, M.; Schneider, Y.J.; Crichton, R.R. Deferoxamine augments growth and pathogenicity of Rhizopus, while hydroxypyridinone chelators have no effect. Kidney Int. 1994, 45, 667–671, doi:10.1038/ki.1994.89.
- Ibrahim, A.S.; Gebermariam, T.; Fu, Y.; Lin, L.; Husseiny, M.I.; French, S.W.; Schwartz, J.; Skory, C.D.; Edwards, J.E.; Spellberg, B.J. The iron chelator deferasirox protects mice from mucormycosis through iron starvation. J. Clin. Invest. 2007, 117, 2649–2657, doi:10.1172/JCI32338.
- Ibrahim, A.S.; Gebremariam, T.; Luo, G.; Fu, Y.; French, S.W.; Edwards, J.E.; Spellberg, B. Combination Therapy of Murine Mucormycosis or Aspergillosis with Iron Chelation, Polyenes, and Echinocandins. Antimicrob. Agents Chemother. 2011, 55, 1768–1770, doi:10.1128/AAC.01577-10.

