 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jaroslav Pejchal | + 4006 word(s) | 4006 | 2020-11-30 07:03:34 | | | |
| 2 | Bruce Ren | Meta information modification | 4006 | 2020-12-08 02:59:39 | | | | |
| 3 | Bruce Ren | Meta information modification | 4006 | 2020-12-24 02:59:44 | | |
Video Upload Options
The organophosphorus substances, including pesticides and nerve agents (NAs), represent highly toxic compounds. Standard decontamination procedures place a heavy burden on the environment. Given their continued utilization or existence, considerable efforts are being made to develop environmentally friendly methods of decontamination and medical countermeasures against their intoxication. Enzymes can offer both environmental and medical applications. One of the most promising enzymes cleaving organophosphorus compounds is the enzyme with enzyme commission number (EC): 3.1.8.2, called diisopropyl fluorophosphatase (DFPase) or organophosphorus acid anhydrolase from Loligo Vulgaris or Alteromonas sp. JD6.5, respectively. Structure, mechanisms of action and substrate profiles are described for both enzymes. Wild-type (WT) enzymes have a catalytic activity against organophosphorus compounds, including G-type nerve agents. Their stereochemical preference aims their activity towards less toxic enantiomers of the chiral phosphorus center found in most chemical warfare agents. Site-direct mutagenesis has systematically improved the active site of the enzyme. These efforts have resulted in the improvement of catalytic activity and have led to the identification of variants that are more effective at detoxifying both G-type and V-type nerve agents. Some of these variants have become part of commercially available decontamination mixtures.
1. Introduction
Biological decomposition of toxic substances has become a very attractive topic. At present, particular emphasis is placed on industrial processes and technologies that do not burden the environment but help to clean or protect it. With increasing intensity and interest in the introduction of “green” industrial technologies, world organizations pursue the reduction of environmental pollution as the result of the increasingly loud calling of society [1]. Biodegradation is a means to decrease toxicity completely without burdening the environment. Biodegradation is defined as a process of decomposing toxic compounds by living organisms without producing other hardly degradable substances. In this regard, enzymatic degradation is a subtype of biodegradation when only enzymes are employed for the degradation of toxic compounds. The utilization of whole living microorganisms poses another approach to biodegradation [2].
Organophosphorus compounds (OP), including pesticides and nerve agents (NA), represent a target for enzymatic degradation. Pesticides are still an integral part of agriculture. Tens of thousands of tons are applied all around the world every year. In the USA, more than 40 thousand tons of OP compounds, including pesticides, are land applied, and 20 thousand tons are produced for export every year [3][4]. Worldwide, OP compounds account for over 38% of the total pesticides used [4][5]. According to the World Health Organization, there are three million pesticide poisonings every year [6]. Another part of this issue is the possibility that these substances leak into the ground and enter municipal water supplies and pollute the surrounding environment.
NA and their potential misuse by a terrorist organization are another current issue. NA are 100–10,000 times more toxic than pesticides. Despite a ban on their use (Chemical Weapons Convention in 1993), there is evidence of misuse by terrorist organizations, extremists or political authoritarianism [7][8]. Sarin was recently used in Ghouta and Khan Shaykhun, Syria, in 2013 and 2017, respectively. Approximately 1400 civilians died in Ghouta [9][10]. S-[2-(diisopropylamino)ethyl]-O-ethyl-methylphosphonothioate (VX), having the abbreviation VX according to NATO designation, was utilized in the assassination of Kim Jong-nam, the half-brother of the North Korean dictator, in Kuala Lumpur, Malaysia in 2017 [11]. In Japan, the extremist group called Aum Shinrikyo applied sarin and VX for their terrorist attack. The most serious of these attacks resulted in the deaths of 19 people and at least 5500 were injured [12][13]. The last abuse of NA was in Salisbury, the United Kingdom, in 2018, when a Novichok agent was used in an attempted assassination [14][15]. Novichok substances were developed in the USSR. Their structures have never been published, but according to the available sources, more than three compounds were synthesised and their toxicities are presumably higher than other NA [16].
Thus, it is clear that OP are a significant health threat and environmental load. Enzymes may significantly facilitate their degradation and support the final decontamination of hot zones. Additionally, pesticide degradation would minimize their harmful effects on the environment. Last but not least, enzymes, which prove to be safe for human intravenous administration, can be utilized as protective as well as therapeutic drugs of both pesticide and NA poisonings [17][18][19].
Naturally occurring enzymes, that hydrolyse organophosphates, act 40 to 2000 times faster than chemical hydrolysis [20]. Therefore, research teams have been investigating the possibilities of biological degradation of chemical warfare agents (CWA) [21]. Several enzymes from different kinds of organisms (bacteria, protozoa, squid, clams, and mammals) have been reported to cleave and detoxify specific groups of pesticides and NA. These enzymes, with varying esterase specificities, have been designated as phosphoric triester hydrolases (EC: 3.1.8) with two subgroups, including aryldialkylphosphatase (EC: 3.1.8.1), also called phosphotriesterase (PTE) or organophosphorus hydrolase (OPH) and diisopropyl fluorophosphatase (DFPase, EC: 3.1.8.2), also called organophosphorus acid anhydrolase (OPAA), tabunase, or organophosphorus anhydrase, etc. [21][22][23][24][25]. PTE isolated from Brevundimonas diminuta GM (formerly Pseudomonas diminuta GM) and Sphingobium fuliginis ATCC 27,551 (formerly Flavobacterium sp. ATCC 27551) have been widely studied.
2. History of EC: 3.1.8.2
Mazur began to explore the possibility of enzymatic degradation of NA shortly after the end of World War II. He identified an enzyme later called DFPase present in a variety of rabbit and human tissues. The enzyme was capable of hydrolysing diisopropyl fluorophosphate (DFP) [26][27][28].
During the 1950s, researchers Mounter, Aldridge and Augustinsson did much of the work in this field [8,26]. Mounter and co-workers [29][30] further investigated and characterized enzymes originally reported by Mazur. They purified the enzyme and determined the effect of metal ions (Co2+, Mn2+) and amino acids on DFPase activity. They also demonstrated the existence of various types of DFPase in up to 14 different forms originating from rats, humans, cats, guinea pigs, pigeons, and turtles [31][32]. This group was also the first one which reported on DFPase from microorganisms [33][34].
Aldridge referred to an enzyme derived from rabbit serum, which was capable of hydrolysing paraoxon. He called it A-esterase or A-serum esterase. He also investigated the stereospecificity of A‑serum esterase for sarin [35][36]. Augustinsson et al. [37] followed up on Aldridge’s research and expanded the work with other enzymes that would hydrolyse CWA, such as tabun. They subsequently showed that the enzyme was more stable at neutral pH and also confirmed Aldridge’s observation that phosphoryl phosphatase isolated from pig kidney exhibits stereoselectivity against tabun [38][39][40]. Due to differences in substrate specificity and sensitivity to inhibition, they distinguished three different types of esterase activity in plasma, including arylesterase (aromatic esterase, A-esterase), aliesterase (carboxylesterase, B-esterase, “lipase”) and cholinesterase. Individual enzymes also displayed variations in their properties [40][41][42].
In the 1960s, many additional scientists focused their attention on the enzymatic hydrolysis of CWA, including Hoskin and his team. They purified and characterized DFPase from squids (Loligo vulgaris, Loligo pealei). The biological and chemical properties of these enzymes are different from all other types of DFPases [43][44][45]. The squid DFPases are highly stabile and possess broad substrate specificity. The molecular weight of the enzyme from Loligo pealei is approximately 30 kDa, requires Ca2+ for its activity and is found only in cephalopods. The gene from Loligo vulgaris was cloned, sequenced, expressed and further characterized [46][47][48]. Its molecular weight is 35.2 kDa. Scharff et al. [49][50] crystallized the enzyme and determined its three-dimensional structure. It has been demonstrated that one of the Ca2+ ions serves to stabilize the structure, while the second one serves the catalytic function. Industrial production (15 kg) of the recombinant DFPase was started by Roche Diagnostic (Berne, Switzerland).
From the early 1980s to the present, researchers have oriented their investigations toward targeted isolation and characterization of microorganisms and their enzymes, and rather a random exploration of organisms having enzymes that hydrolyse CWA, especially nerve agents. Enzymes with DFPase activity were discovered in the ciliate protozoan Tetrahymena thermophilia by Landis et al. [51][52][53] or in the clam Rangia cuneata by Anderson et al. [54][55]. These enzymes have a different molecular weight, hydrolysis rate, substrate specificity, and metal stimulation. Attaway and co-workers [56]creened 18 g-negative bacterial isolates and observed that only cultures with parathion hydrolase activity showed significant DFPase levels.
As the nomenclature of these enzymes was unsystematic and confusing, the literature soon became filled with references to phosphorylphosphatase, fluorophosphatase, DFPase, paraoxon, parathion hydrolase, phosphotriesterase, phosphofluorase, somanase, sarinase, and tabunase. The Nomenclature Committee of the International Union of Biochemistry and Molecular Biology established a new systematic nomenclature in 1992. Currently, two main types of DFPases are used to degrade CWA. The first DFPase is isolated from squids (also known as squid-type DFPases) with a molecular weight of 30–40 kDa and the second one is from bacteria Alteromonas species (also known as Mazur-type DFPases or OPAA) with a molecular weight of 40–96 kDa. These two enzymes are the most studied [57]. A general scheme of degradation of organophosphorus compounds by DFPase and OPAA is presented in Scheme 1.
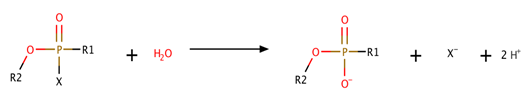
Scheme 1. Schematic degradation of organophosphorus compounds by diisopropyl fluorophosphatase (DFPase) and organophosphorus acid anhydrolase (OPAA).
3. Squid-Type DFPase
Squid-type DFPase is the oldest known enzyme utilized for the decontamination of organophosphorus compounds. However, it is still relevant for large-scale production . It is quite remarkable that an enzyme hydrolysing highly toxic OP was found in a higher organism. The discovery of DFPase is linked with the research by Nachmansohn and Hoskin. Nachmansohn’s group [58] used the squid Loligo pealei for their experiments. In one of them, they tried to block axonal conduction by irreversibly inhibiting the cholinesterases using the potent inhibitor DFP. However, the DFP concentration needed to suppress the conduction turned out to be exceptionally high. Hoskin et al. [59] tried to investigate this surprising behaviour. They concluded that the axonal envelope contains a potent hydrolytic enzyme. They continued this work and isolated this enzyme in the hepatopancreas, saliva, head ganglion, and axons [60][61]. They also demonstrated that the enzyme is able to hydrolyse sarin, soman and tabun.
3.1. Structure
The structural investigation began in parallel with studies of the chemical effect and kinetic studies [47]. The first X-ray structure was solved at a resolution of 1.8 Å (PDB—code 1E1A). Another determination of the DFPase structure was able to push the resolution to 0.85 Å (PDB—code 1PJX) [62]. This improvement caused better imaging of the enzyme structure and an understanding of its mechanism of action. There are some similarities between L. vulgaris and the other squid‑type DFPases. Squid-type DFPases were found to be metal-dependent hydrolases with calcium ions (Ca2+) as the native metal, although manganese (Mn2+) can be substituted for the calcium ions [63][64][65][66][67]. DFPase isolated from the head ganglion of L. vulgaris has a monomeric structure containing 314 amino acids in the 35 kDa polypeptide chain. The enzyme has a β-propeller structure with six (propeller) blades. Each blade consists of 4-stranded antiparallel β-sheets. They are arranged in a 6-fold pseudosymmetry around a central water-filled tunnel that contains both calcium ions [66]. More solvent-exposed calcium ion has been identified as the “low-affinity” calcium (Ca-1). Ca-1 is located at the base of the active site, sealing the water-filled tunnel and playing a critical role in the enzymatic activity. The more buried calcium ion is known as “high-affinity” calcium (Ca-2). Ca-2 is in the center of the molecule and is responsible for the structural integrity of the enzyme [68][69]. Calcium ions are bound to the enzyme by amino acid side chains and the loss of active site-bound ion destroys the enzyme activity (Figure 1) [69]. The active site analysis shows that the calcium ions are coordinated by four amino acid residues at the bottom of the active site. The three remaining ligands are water molecules. Two of them are below the metal ion forming the “dead end” of the central water-filled tunnel and one is on the top of the metal ion in the active site. Biophysical and site-directed mutagenesis studies have indicated that one particular histidine residue, His287, is important but not essential for the hydrolytic reaction. Hartleib and Rüterjans replaced His287 with Asn, producing a mutant protein with only 3.7% residual enzymatic activity. According to the X-ray structure, both His287 and Ca-1 are located in a solvent-accessible surface pocket of DFPase. Structural and catalytic types are also ligated to the protein by Asp232, His274 and by Glu21, Asn120, Asn175, and Asp229 side chains, respectively [68]. Particularly, the catalytic residues have been comprehensively mutated. The studies showed that calcium coordination is dependent on the presence of Glu21 and at least two negatively charged residues in the active site [70][71]. Although calcium binding is crucial, its presence is not sufficient for enzymatic activity. The active site’s overall charge and the electrostatic topology are also critical [70], as single mutants of Asn120 and Asn175 contain calcium but display only 4% and 2% residual enzymatic activity, respectively [69].
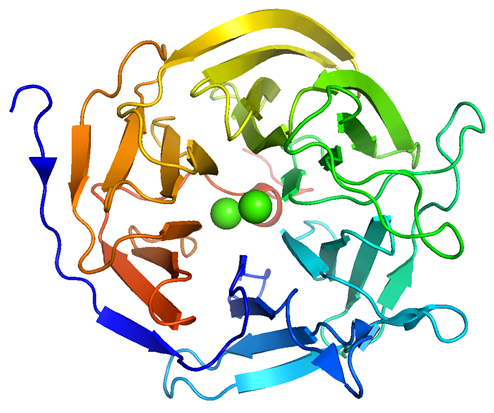
Figure 1. DFPase from Loligo vulgaris (PDB ID 3O4P, resolution 0.85 Å) [72]: six foursomes of antiparallel β-sheets (six blades) create the central tunnel containing two calcium ions (green); coloured from the N-terminal (blue) to C-terminal (red).
3.2. Mechanism of Action
Two possible catalytic mechanisms have been described. The structure of the active site of the enzyme is shown in Figure 2. The first mechanism and X-ray structure were studied at the same time, and investigation assumes the essential role of one particular histidine residue (His287) for the hydrolytic reaction. Based on the X-ray structure, both His287 and Ca-1 are located in a solvent-accessible surface pocket of DFPase and His287 and Trp244 are connected by a hydrogen bridge. His287 also forms another hydrogen bond with the backbone carbonyl oxygen of Ala20. This arrangement is unable to activate the hydrolytic water molecule; therefore, a structural rearrangement in the active site is necessary for the enzymatic cleavage reaction. According to this mechanism, the incoming substrate is replaced by a calcium coordinating water molecule in the active site. It increases the partial positive charge of the phosphorus atom, which facilitates the nucleophilic attack of the hydrolytic water molecule. This water molecule may be activated via proton abstraction by His287 when the imidazole ring is disengaged from the hydrogen bond interactions with Trp244 and Ala20. At this point, the double-protonated His287 side chain interacts with the negatively charged Glu37. Metal (Ca2+ ion) has a function of electrophilic Lewis acid and it also helps the nucleophilic reaction. All of these interactions lead to a nucleophilic attack of the subsequently released hydroxyl ion on the phosphorus atom of the bound substrate. Finally, the fluoride ion is cleaved and released in the opposite direction from the attacking water. After deprotonation of the charged His287, the hydrogen bond interaction with Trp244 can be restored to initiate a new reaction cycle (Scheme 2). The function of the central tunnel has not been revealed to date. However, this reaction mechanism was questioned when DFPase mutations (His287Phe and His287Leu) were generated and retained 65–80% activity of the wild-type enzyme.
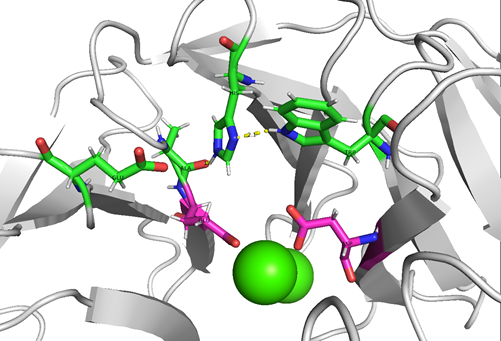
Figure 2. DFPase from Loligo vulgaris (PDB ID 3O4P, resolution 0.85 Å) [72]: a detailed view of the two possible catalytic residues: (1) Ala20, Glu37, Trp244, and His287 coloured green and (2) Glu21 and Asp229 coloured magenta.
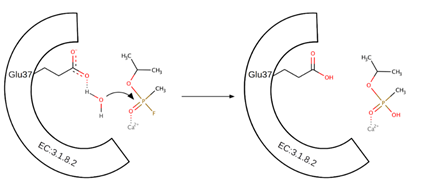
Scheme 2. Mechanism of nucleophilic substitution (SN2) of the fluorine group to the hydroxyl group in sarin. Glu37 activates a water molecule. The released hydroxyl ion attacks the partially charged phosphorus atom.
A new, and more probable mechanism, was proposed based on new experimental findings. Computational simulation studies have suggested an essential role of the Asp229 residue in the DFPase catalytic activity because of its correct orientation for performing nucleophilic attack in the hydrolysis mechanism through bimolecular nucleophilic substitution (SN2). Asp229 is also coordinated to the Ca2+ ion, together with other residues and water molecules. There are two possible pathways of how the reaction can proceed. The first pathway for catalytic hydrolysis refers to Asp229 as a nucleophile, attacking the phosphorus center of the substrate coordinated to the Ca2+ ion. Pentavalent phosphoenzyme intermediate is formed and subsequently, the intermediate is hydrolysed, with the assistance of Glu21, by water attacking the carboxylated carbon atom of Asp229. This leads to the regeneration of the enzyme and the release of the product (Scheme 3) [73][74][75][76]. This mechanism is also in concordance with Blum et al. [77]. In one of their studies, DFPase cleaved DFP in 18O-labelled water. An excessive amount of enzyme was used to simulate single-turnover conditions, whereas an excess of the substrate was utilized for multiple-turnover reactions. Single-turnover experiments demonstrated the incorporation of 16O and thus revealed that the reaction proceeds via a phosphoenzyme intermediate with enzyme being the only source of 16O. Subsequent mass spectrometry analysis identified an oligopeptide with an altered mass, corresponding to the fragment containing Asp229 [74]. Another study employed neutron diffraction to show the deprotonation of Asp229, supporting its function as a potential nucleophile [77].
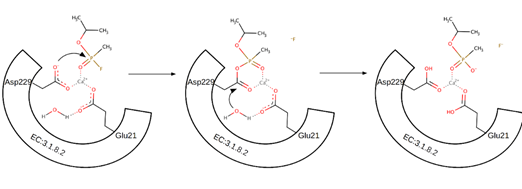
Scheme 3. Mechanism of two-step nucleophilic substitution of the fluorine group to the hydroxyl group in sarin. In the first step, the Asp229 attacks the phosphorous atom, releasing the fluorine ion. Glu21 then activates the water molecule. The released hydroxyl ion finally attacks the carbonyl carbon atom of Asp229. The product is released and the enzyme is regenerated.
The second pathway was described based on (S)-sarin computational simulations [76]. The pathway points to the participation of a water molecule as a nucleophile with the assistance of both Asp229 and Glu21 residues. The OP substrate is also coordinated to the Ca2+ ion via phosphoryl oxygen. The water molecule is activated by proton abstraction through Asp229, and the hydroxyl ion is formed. This ion acts as a nucleophile and attacks the phosphoric center of the substrate. It leads to the pentavalent intermediate. The Glu21 participates in these proton transfers. Finally, the leaving group is released and the hydrolysed substrate is removed from the Ca2+ ion at the ionized form as the reaction product (Scheme 4). Notably, although many studies on the mechanism of action exist, there is no consensus in the literature describing the exact reaction pathway.
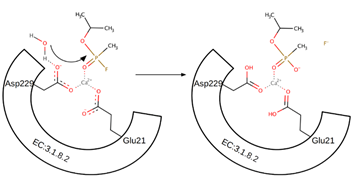
Scheme 4. Mechanism of nucleophilic substitution (SN2) of the fluorine group to the hydroxyl group in sarin. The negatively charged amino acid residues activate the water molecule. The hydroxyl ion is a nucleophile attacking the partially charged phosphorus atom.
3.3. Substrate Profile
At present, the physiological role of the enzyme remains unknown and no natural substrate for DFPase has yet been identified . However, the substrate profile of this enzyme is quite broad. Squid DFPases can hydrolyse organophosphorus substrates with a P-F or P-CN bond, including sarin, cyclosarin, soman and tabun [78][79]. However, it cannot detoxify agents with P-O or P-S bonds, such as paraoxon, VX or N,N-diethyl-2-(methyl-(2-methylpropoxy)phosphoryl)sulfanyl- ethanamine (known as Russian VX or VR according to the NATO designation). Catalytic efficiency for these NA is less than that for DFP, one-tenth for tabun and approximately one-third for the others. Squid DFPases are characterized by stereoselectivity towards the less toxic Sp-enantiomer of organophosphorus compounds, yet complete detoxification by wild-type enzyme can be achieved. The enzyme has some additional advantages. DFPase is a highly stable protein, displaying activity over a wide temperature and pH range. It is also compatible with large amounts of different organic solvents and microemulsions as carrier systems [79].
4. OPAA from Alteromonas
In the 1980s, OPAA enzymes were identified in halophilic bacteria called Alteromonas . The most studied strain was Alteromonas sp. JD6.5, which will be described in more detail in this article. However, other strains, like Alteromonas haloplanktis or Alteromonas undina, produce the enzyme capable of hydrolysing organophosphorus compounds as well. All these OPAAs are structurally and functionally similar to each other. They share a molecular weight between 50–60 kDa, an optimum pH from 7.5 to 8.5, an optimum temperature range from 40 to 55 °C and all require an Mn2+ ion for maximum activity. See Table 1 for specific values [80][81][82][83][84].
Table 1. Comparison of various Alteromonas OPAAs.
|
- |
Alteromonas sp. JD6.5 |
Alteromonas haloplanktis |
Alteromonas undina |
|
Molecular weight (kDa) |
60 |
50 |
53 |
|
Metal requirement |
Mn or Co |
Mn |
Mn |
|
Temperature optimum (°C) |
50 |
40 |
55 |
|
pH optimum |
8.5 |
7.5 |
8.0 |
4.1. Structure
Bacterial isolate from Alteromonas sp. JD6.5 was obtained from water and soil samples of salt springs near the Great Salt Lake in the state of Utah. It is a gram-negative, aerobic short rod and needs at least 2% NaCl for growing, with an optimum between 5–10% NaCl [8,22,80]. At present, there is still little knowledge of the structure and mechanism of action of this enzyme. The native enzyme’s structure was solved at 2.3 Å resolution [85]. OPAA is a single polypeptide and is composed of 517 amino acids. A very similar enzyme was isolated from Alteromonas haloplanktis containing 440 amino acids. Both enzymes have 77–80% amino acid homology. The remaining invisible 77-residue segment from Alteromonas sp. JD6.5 has high flexibility. OPAA from Alteromonas sp. JD6.5 is also classified as prolidase based on its structure and catalytic properties. OPAA was firstly identified as a monomer, but now after further experiments, it has been determined as a tetramer [86][87].
OPAA structure consists of two domains, a small N-terminal domain and a large C-terminal domain, harbouring the binuclear Mn2+ ions in the active site (Figure 3) [88]. Six β‑sheets are present in the N-terminal domain with an antiparallel central pair of strands and parallel flanking pairs. On the other hand, all five β‑strands in the C-domain are antiparallel. The β-sheet in the N-domain is twisted, whereas that in the C-domain is strongly curved with the exhibiting “pita bread” architecture [85]. There are some differences in contents and topological arrangements of α-helices in the two domains. The N-domain contains four helices, while the C-domain with the pita bread architecture contains eight helices [85]. The binuclear metal center is located in the C-terminal region, which consists of the amino acid residues Asp244, Asp255, His336, Glu381, and Glu420 [85]. Two bridging Mn2+ ions are needed for full catalytic activity. Also, a nonproteinaceous density was found in the active site of the enzyme, however, not definitively determined. It is assumed to be a bonded glycolate whose three oxygen atoms coordinate the two Mn2+ ions [85].
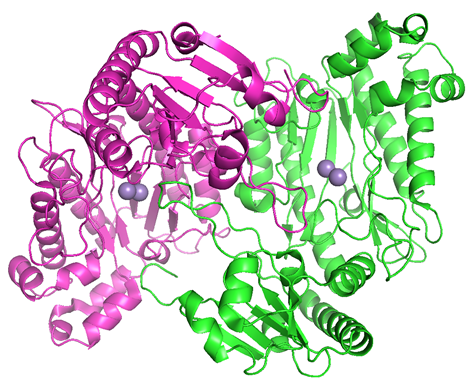
Figure 3. Organophosphate anhydrolase/prolidase from Alteromonas sp. (PDB ID 4ZWO, mutant Y212F, resolution 2.14 Å) [88]: the two chains of the homo-dimer are coloured green and magenta; manganese ions are coloured violet.
4.2. Mechanism of Action
The substrate-binding site in OPAA is comprised of three pockets (large, small, and the leaving group pocket) as other similar enzymes. The large pocket is formed by Leu225, His226, His332, and Arg418. The small pocket is composed of Tyr212, Val342, His343, and Asp45 from the N-terminal domain of the opposite subunit in the dimer. The leaving group pocket is formed by residues Tyr292 and Leu366. (Figure 4). The ability of this enzyme to cleave OP compounds, including G-type NA and proline dipeptides, comes from the bridging water molecule or hydroxide ion in the metal center that helps to promote a nucleophilic attack on either the carbonyl oxygen of the scissile peptide bond of the dipeptide [Xaa-Pro] or the phosphorus center of NA.
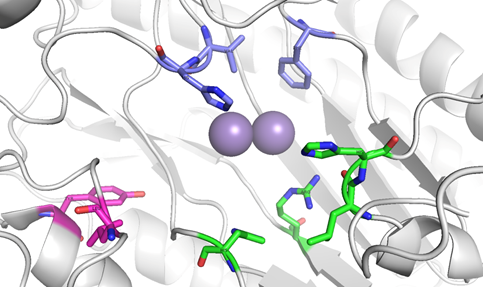
Figure 4. Organophosphate anhydrolase/prolidase from Alteromonas sp. (PDB ID 4ZWO, mutant Y212F, resolution 2.14 Å): the large pocket formed by Leu225, His226, His332 and Arg418 (coloured green), the leaving group pocket formed by Tyr292 and Leu366 (coloured magenta), and the small pocket formed by Tyr212, Val342, His343, and Asp45 from the opposite chain (coloured blue).
4.3. Substrate Profile
OPAA belongs to a class of bimetalloenzymes that can hydrolyse various toxic OP compounds. It can also be classified as a prolidase cleaving dipeptides with proline in the C-terminus. Due to this aspect, the enzyme’s physiological role might be associated with cellular dipeptide metabolism [89]. A high level of hydrolysis activity was observed against OP compounds with P-F bonds, but very minimal activity for P-O or P-C bonds and no activity against P-S bonds. Similar substrate activity was later identified for the OPAA from Alteromonas undina and Alteromonas haloplanktis (Table 2). The best substrate for OPAA hydrolysis is soman. However, OPAA also catalyses the hydrolysis of other NA like sarin, tabun, diisopropyl fluorophosphate, and nerve agents analogues containing a p‑nitrophenol leaving group. The enzyme less effectively hydrolyses novichok substances. On the other hand, minimal catalytic activity was observed for V-type nerve agents (Table 3) [8,21,22,81,86,90–92] and mipafox (N,N′-diisopropyl phosphorodiamidofluoridate, DDFP), a close DFP analogue, is not a substrate under normal assay conditions but a competitive inhibitor. In general, OPAA has a preference for the less toxic stereoisomer. The OPAA stereoselectivity is for the Rp-enantiomers of methyl phosphonates with a selectivity of higher than 7000-fold to analogues of soman [90].
Table 2. Substrate specific activity (U.mg−1) of various Alteromonas OPAAs.
|
Agent/Source |
Alteromonas undina |
Alteromonas haloplanktis |
Alteromonas sp. JD6.5 |
|
DFP |
1403 |
691 |
1820 |
|
tabun |
368 |
255 |
85 |
|
sarin |
426 |
308 |
611 |
|
soman |
2826 |
1667 |
3145 |
|
cyclosarin |
1775 |
323 |
1654 |
Table 3. Organophosphorus substrate specificity for Alteromonas sp. JD6.5 OPAA [87,88,92,93].
|
Substrate |
kcat/Km (M−1.min−1) |
|
DFP |
3.7 × 107 |
|
soman |
1.6 × 107 |
|
GP |
1.3 × 107 |
|
sarin |
1.3 × 106 |
|
novichok A230 |
9.5 × 104 |
|
novichok A232 |
6.9 × 104 |
|
novichok A234 |
3.5 × 104 |
|
VR |
5.4 × 102 |
References
- Linthorst, J.A. An overview: origins and development of green chemistry. Found. Chem. 2009, 12, 55–68, doi:10.1007/s10698-009-9079-4.
- Dvořák, J.; Melkes, V. Ekologické havárie a dekontaminace znečištění; Vysoká vojenská škola pozemního vojska: Vyškov, The Czech Republic, 1997.
- Mulchandani, A.; Kaneva, I.; Chen, W. Detoxification of organophosphate nerve agents by immobilizedEscherichia coli with surface-expressed organophosphorus hydrolase. Biotechnol. Bioeng. 1999, 63, 216–223, doi:10.1002/(sici)1097-0290(19990420)63:23.0.co;2-0.
- Ghanem, E.; Raushel, F.M. Detoxification of organophosphate nerve agents by bacterial phosphotriesterase. Toxicol. Appl. Pharmacol. 2005, 207, 459–470, doi:10.1016/j.taap.2005.02.025.
- Singh, B.K.; Walker, A. Microbial degradation of organophosphorus compounds. FEMS Microbiol. Rev. 2006, 30, 428–471, doi:10.1111/j.1574-6976.2006.00018.x.
- WHO. Public Health Impact of Pesticides used in Agriculture; World Health Organization: Geneva, Switzerland, 1990.
- Las, B.R. Southeast Asia: A Potential Domain for Chemical Terrorism; Missouri State University: Springfield, MA, USA, 2019.
- DeFrank, J. Catalytic Enzyme-Based Methods for Water Treatment and Water Distribution System Decontamination; U.S. Army Edgewood Chemical Biological Center, Aberdeen Proving Ground: Gunpowder, MD, USA, 2006.
- UN Security Council. Report of the OPCW Fact-Finding Mission in Syria Regarding an Alleged Incident in Khan Shaykhun, Syrian Arab Republic April 2017 (S/2017/567) [EN/AR], 2017. Available online: https://reliefweb.int/report/syrian-arab-republic/report-opcw-fact-finding-mission-syria-regarding-alleged-incident-khan (accessed on 1 August 2020).
- Rosman, Y.; Eisenkraft, A.; Milk, N.; Shiyovich, A.; Ophir, N.; Shrot, S.; Kreiss, Y.; Kassirer, M. Lessons Learned From the Syrian Sarin Attack: Evaluation of a Clinical Syndrome Through Social Media. Ann. Intern. Med. 2014, 160, 644–648, doi:10.7326/m13-2799.
- BBC News. Kim Jong-nam ’Killed by VX Nerve Agent’. 2017. Available online: https://www.bbc.com/news/world-asia-39073389 (accessed on 1 August 2020).
- OPCW. The Sarin Gas Attack in Japan and the Related Forensic Investigation. Available online: https://www.opcw.org/media-centre/news/2001/06/sarin-gas-attack-japan-and-related-forensic-investigation, 2001 (accessed on 1 August 2020).
- Bigley, A.N.; Raushel, F.M. The evolution of phosphotriesterase for decontamination and detoxification of organophosphorus chemical warfare agents. Chem. Interact. 2019, 308, 80–88, doi:10.1016/j.cbi.2019.05.023.
- OPCW. Incident in Salisbury. Available online: https://www.opcw.org/media-centre/featured-topics/incident-salisbury, 2018 (accessed on 1 August 2020).
- Peplow, M. Nerve Agent Attack on Spy Used ‘Novichok’ Poison. 2018 Available online: https://cen.acs.org/articles/96/i12/Nerve-agent-attack-on-spy-used-Novichok-poison.html (accessed on 1 August 2020).
- Nepovimova, E.; Kuča, K. Chemical warfare agent NOVICHOK-mini-review of available data. Food Chem. Toxicol. 2018, 121, 343–350, doi:10.1016/j.fct.2018.09.015.
- Chen, W.; Richins, R.D.; Mulchandani, P.; Kaneva, I.; Mulchandani, A. Biodegradation of organophosphorus nerve agents by surface expressed organophosphorus hydrolase. In Enzymes in Action; Springer Science and Business Media LLC: Dordrecht, The Netherlands, 2000; pp. 211–221.
- Singh, B.K. Organophosphorus-degrading bacteria: ecology and industrial applications. Nat. Rev. Genet. 2009, 7, 156–164, doi:10.1038/nrmicro2050.
- Hoskin, F.C.; Walker, J.E.; Stote, R. Degradation of nerve gases by CLECS and cells: kinetics of heterogenous systems. Chem. Interact. 1999, 119, 439–444, doi:10.1016/s0009-2797(99)00056-3.
- Schofield, D.A.; DiNovo, A. Generation of a mutagenized organophosphorus hydrolase for the biodegradation of the organophosphate pesticides malathion and demeton-S. J. Appl. Microbiol. 2010, 109, 548–557, doi:10.1111/j.1365-2672.2010.04672.x.
- Raushel, F.M. Bacterial detoxification of organophosphate nerve agents. Curr. Opin. Microbiol. 2002, 5, 288–295, doi:10.1016/s1369-5274(02)00314-4.
- Theriot, C.M.; Grunden, A.M. Hydrolysis of organophosphorus compounds by microbial enzymes. Appl. Microbiol. Biotechnol. 2011, 89, 35–43, doi:10.1007/s00253-010-2807-9.
- Richardt, A.; Blum, M.-M. Decontamination of Warfare Agents; Wiley-VCH: Weinheim, Germany, 2008.
- Dave, K.I.; Miller, C.E.; Wild, J.R. Characterization of organophosphorus hydrolases and the genetic manipulation of the phosphotriesterase from Pseudomonas diminuta. Chem. Interact. 1993, 87, 55–68, doi:10.1016/0009-2797(93)90025-t.
- DeFrank, J.J.; White, W.E. Phosphofluoridates: Biological Activity and Biodegradation; Springer Science and Business Media LLC: Dordrecht, The Netherlands, 2006; pp. 295–343.
- Prokop, Z.; Opluštil, F.; DeFrank, J.; Damborsky, J. Enzymes fight chemical weapons. Biotechnol. J. 2006, 1, 1370–1380, doi:10.1002/biot.200600166.
- Mazur, A. An enzyme in animal tissues capable of hydrolyzing the phosphorus-fluorine bond of alkyl fluorophosphates. J. Biol. Chem. 1946, 164, 271–289.
- Clancy, J.; McVicar, A.; Ellison, D.H. Handbook of Chemical and Biological Warfare Agents; Informa UK Limited: Colchester, UK, 2007.
- Mounter, L.A.; Floyd, C.S.; Chanutin, A. Dialkylfluorophosphatase of kidney. I. Purification and properties. J. Biol. Chem. 1953, 204, 221–232.
- Mounter, L.A. The complex nature of dialkylfluorophosphatases of hog and rat liver and kidney. J. Biol. Chem. 1955, 215, 705–711.
- Mounter, L.A.; Dien, L.T.; Chanutin, A. The distribution of dialkylfluorophosphatases in the tissues of various species. J. Biol. Chem. 1955, 215, 691–697.
- Mounter, L.A.; Dien, L.T. Dialkylfluorophosphatase of kidney. V. The hydrolysis of organophosphorus compounds. J. Biol. Chem. 1956, 219, 685–690.
- Mounter, L.A.; Tuck, K.D. Dialkylfluorophosphatases of microorganisms. II. Substrate specificity studies. J. Biol. Chem. 1956, 221, 537–541.
- Mounter, L.A.; Baxter, R.F.; Chanutin, A. Dialkylfluorophosphatases of microorganisms. J. Biol. Chem. 1955, 215, 699–704.
- Aldridge, W.N. Serum esterases. 1. Two types of esterase (A and B) hydrolysing p-nitrophenyl acetate, propionate and butyrate, and a method for their determination. Biochem. J. 1953, 53, 110–117, doi:10.1042/bj0530110.
- Aldridge, W.N. Serum esterases. 2. An enzyme hydrolysing diethyl p-nitrophenyl phosphate (E 600) and its identity with the A-esterase of mammalian sera. Biochem. J. 1953, 53, 117–124, doi:10.1042/bj0530117.
- Augustinsson, K.-B.; Heimbürger, G.; Roine, P.; Sörensen, N.A. Enzymic Hydrolysis of Organophosphorus Compounds. I. Occurrence of Enzymes Hydrolyzing Dimethyl-amido-ethoxy-phosphoryl Cyanide (Tabun). Acta Chem. Scand. 1954, 8, 753–761, doi:10.3891/acta.chem.scand.08-0753.
- Augustinsson, K.-B.; Heimbürger, G.; Risberg, E.; Lamm, O. Enzymatic Hydrolysis of Organophosphorus Compounds. VI. Effect of Metallic Ions on the Phosphorylphosphatases of Human Serum and Swine Kidney. Acta Chem. Scand. 1955, 9, 383–392, doi:10.3891/acta.chem.scand.09-0383.
- Augustinsson, K.-B.; Studnitz, W.V.; Bergson, G.; Grönvall, A.; Zaar, B.; Diczfalusy, E. Enzymic Hydrolysis of Organophosphorus Compounds. VIII. Effect of Anions. Acta Chem. Scand. 1958, 12, 1286–1291, doi:10.3891/acta.chem.scand.12-1286.
- Augustinsson, K.-B.; Kulonen, E.; Hevesy, G.; Schliack, J.; Reio, L. Enzymatic Hydrolysis of Organophosphorus Compounds. VII. The Stereospecificity of Phosphorylphosphatases. Acta Chem. Scand. 1957, 11, 1371–1377, doi:10.3891/acta.chem.scand.11-1371.
- Augustinsson, K.-B.; Heimbürger, G.; Vihovde, J.; Sörensen, N.A. Enzymic Hydrolysis of Organophosphorus Compounds. IV. Specificity Studies. Acta Chem. Scand. 1954, 8, 1533–1541, doi:10.3891/acta.chem.scand.08-1533.
- Augustinsson, K.-B.; Heimbürger, G.; Ingri, N.; Lamm, O. Enzymatic Hydrolysis of Organophosphorus Compounds. V. Effect of Phosphorylphosphatase on the Inactivation of Cholinesterases by Organophosphorus Compounds in vitro. Acta Chem. Scand. 1955, 9, 310–318, doi:10.3891/acta.chem.scand.09-0310.
- Hoskin, F.C.G. Possible Significance of “DFPase“ in Squid Nerve. Biol. Bull. Mar. Biol. Lab. 1969, 137, 389–390.
- Hoskin, F.C.G. Diisopropylphosphorofluoridate and Tabun: Enzymatic Hydrolysis and Nerve Function. Science 1971, 172, 1243–1245, doi:10.1126/science.172.3989.1243.
- Hoskin, F.C.; Roush, A.H. Hydrolysis of nerve gas by squid-type diisopropyl phosphorofluoridate hydrolyzing enzyme on agarose resin. Science 1982, 215, 1255–1257, doi:10.1126/science.7058344.
- Hartleib, J.; Rüterjans, H. High-Yield Expression, Purification, and Characterization of the Recombinant Diisopropylfluorophosphatase from Loligo vulgaris. Protein Expr. Purif. 2001, 21, 210–219, doi:10.1006/prep.2000.1360.
- Hartleib, J.; Rüterjans, H. Insights into the reaction mechanism of the diisopropyl fluorophosphatase from Loligo vulgaris by means of kinetic studies, chemical modification and site-directed mutagenesis. Biochim. Biophys. Acta BBA Protein Struct. Mol. Enzym. 2001, 1546, 312–324, doi:10.1016/s0167-4838(01)00153-4.
- Hartleib, J.; Geschwindner, S.; Scharff, E.I.; Rüterjans, H. Role of calcium ions in the structure and function of thedi-isopropylfluorophosphatase from Loligo vulgaris. Biochem. J. 2001, 353, 579, doi:10.1042/0264-6021:3530579.
- Scharff, E.I.; Lücke, C.; Fritzsch, G.; Koepke, J.; Hartleib, J.; Dierl, S.; Rüterjans, H. Crystallization and preliminary X-ray crystallographic analysis of DFPase from Loligo vulgaris. Acta Crystallogr. Sect. D Biol. Crystallogr. 2001, 57, 148–149, doi:10.1107/s0907444900014232.
- Scharff, E.I.; Koepke, J.; Fritzsch, G.; Lücke, C.; Rüterjans, H. Crystal structure of diisopropylfluorophosphatase from Loligo vulgaris. Structure 2001, 9, 493–502, doi:10.1016/s0969-2126(01)00610-4.
- Landis, W.G.; Savage, R.E.; Hoskin, F.C.G. An Organofluorophosphate-Hydrolyzing Activity inTetrahymena thermophila1. J. Protozool. 1985, 32, 517–519, doi:10.1111/j.1550-7408.1985.tb04053.x.
- Landis, W.G.; Haley, M.V.; Johnson, D.W. Kinetics of the DFPase Activity inTetrahymena thermophila1. J. Protozool. 1986, 33, 216–218, doi:10.1111/j.1550-7408.1986.tb05593.x.
- Landis, W.G.; Haley, D.M.; Haley, M.V.; Johnson, D.W.; Durst, H.D.; Savage, R.E. Discovery of multiple organofluorophosphate hydrolyzing activities in the protozoanTetrahymena thermophila. J. Appl. Toxicol. 1987, 7, 35–41, doi:10.1002/jat.2550070107.
- Anderson, R.; Durst, H.; Landis, W. Organofluorophosphate-hydrolyzing activity in an estuarine clam, Rangia cuneata. Comp. Biochem. Physiol. Part C Comp. Pharmacol. 1988, 91, 575–578, doi:10.1016/0742-8413(88)90080-1.
- Anderson, R.S.; Durst, H.D.; Landis, W.G. Initial characterization of an OPA anhydrase in the clam, Rangia cuneata. Comp. Biochem. Phys. 1988, 91C, 575–578.
- Attaway, H.; O Nelson, J.; Baya, A.M.; Voll, M.J.; E White, W.; Grimes, D.J.; Colwell, R.R. Bacterial detoxification of diisopropyl fluorophosphate. Appl. Environ. Microbiol. 1987, 53, 1685–1689, doi:10.1128/aem.53.7.1685-1689.1987.
- Hoskin, F.C.G.; Kirkish, M.A.; Steinmann, K.E. Two Enzymes for the Detoxicatlon of Organophosphorus Compounds—Sources, Similarities, and Significance. Toxicol. Sci. 1984, 4, 165–172, doi:10.1093/toxsci/4.2part2.165.
- Nachmansohn, D.; Neumann, E. Chemical and Molecular Basis of Nerve Activity; Elsevier: Amsterdam, The Netherlands, 1975.
- Hoskin, F.C.; Rosenberg, P.; Brzin, M. Re-examination of the effect of DFP on electrical and cholinesterase activity of squid giant axon. Proc. Natl. Acad. Sci. USA 1966, 55, 1231–1235, doi:10.1073/pnas.55.5.1231.
- Hoskin, F.C.; Long, R.J. Purification of a DFP-hydrolyzing enzyme from squid head ganglion. Arch. Biochem. Biophys. 1972, 150, 548–555, doi:10.1016/0003-9861(72)90073-2.
- Garden, J.M.; Hause, S.K.; Hoskin, F.C.; Roush, A.H. Comparison of DFP-hydrolyzing enzyme purified from head ganglion and hepatopancreas of squid (Loligo pealei) by means of isoelectric focusing. Comp. Biochem. Physiol. Part C Comp. Pharmacol. 1975, 52, 95–98, doi:10.1016/0306-4492(75)90020-9.
- Koepke, J.; Scharff, E.I.; Lücke, C.; Rüterjans, H.; Fritzsch, G. Statistical analysis of crystallographic data obtained from squid ganglion DFPase at 0.85 A resolution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2003, 59, 1744–1754, doi:10.1107/s0907444903016135.
- Melzer, M.; Chen, J.C.-H.; Heidenreich, A.; GäbJ.; Koller, M.; Kehe, K.; Blum, M.-M. Reversed Enantioselectivity of Diisopropyl Fluorophosphatase against Organophosphorus Nerve Agents by Rational Design. J. Am. Chem. Soc. 2009, 131, 17226–17232, doi:10.1021/ja905444g.
- Cohen, J.; Warringa, M. Purification and properties of dialkylfluorophosphatase. Biochim. Biophys. Acta BBA Bioenerg. 1957, 26, 29–39, doi:10.1016/0006-3002(57)90050-1.
- Deschamps, J.R.; Kopec-Smyth, K.; Poppino, J.L.; Futrovsky, S.L.; Ward, K. Comparison of organophosphorous acid anhydrolases from different species using monoclonal antibodies. Comp. Biochem. Physiol. Part C: Pharmacol. Toxicol. Endocrinol. 1993, 106, 765–768, doi:10.1016/0742-8413(93)90240-l.
- Allahyari, H.; Latifi, A.M. Diisopropyl-fluorophosphatase as a catalytic bioscavenger. Appl. Biotechnol. Rep. 2016, 3, 477–482.
- Chemnitius, J.-M.; Losch, H.; Losch, K.; Zech, R. Organophosphate detoxicating hydrolases in different vertebrate species. Comp. Biochem. Physiol. Part C Comp. Pharmacol. 1983, 76, 85–93, doi:10.1016/0742-8413(83)90048-8.
- Bigley, A.N.; Raushel, F.M. Catalytic mechanisms for phosphotriesterases. Biochim. Biophys. Acta BBA Proteins Proteom. 2013, 1834, 443–453, doi:10.1016/j.bbapap.2012.04.004.
- Katsemi, V.; Lücke, C.; Koepke, J.; Lohr, F.; Maurer, S.; Fritzsch, G.; Rüterjans, H. Mutational and Structural Studies of the Diisopropylfluorophosphatase fromLoligo vulgarisShed New Light on the Catalytic Mechanism of the Enzyme†. Biochemistry 2005, 44, 9022–9033, doi:10.1021/bi0500675.
- Blum, M.-M.; Chen, J.C.-H. Structural characterization of the catalytic calcium-binding site in diisopropyl fluorophosphatase (DFPase)—Comparison with related β-propeller enzymes. Chem. Interact. 2010, 187, 373–379, doi:10.1016/j.cbi.2010.02.043.
- Chen, J.C.-H.; Mustyakimov, M.; Schoenborn, B.P.; Langan, P.; Blum, M.-M. Neutron structure and mechanistic studies of diisopropyl fluorophosphatase (DFPase). Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 1131–1138, doi:10.1107/s0907444910034013.
- Elias, M.; Liebschner, D.; Koepke, J.; LeComte, C.; Guillot, B.; Jelsch, C.; Chabriere, E. Hydrogen atoms in protein structures: high-resolution X-ray diffraction structure of the DFPase. BMC Res. Notes 2013, 6, 308, doi:10.1186/1756-0500-6-308.
- Xu, C.; Yang, L.; Yu, J.-G.; Liao, R.-Z. What roles do the residue Asp229 and the coordination variation of calcium play of the reaction mechanism of the diisopropyl-fluorophosphatase? A DFT investigation. Theor. Chem. Acc. 2016, 135, 138, doi:10.1007/s00214-016-1896-7.
- Blum, M.-M.; Lohr, F.; Richardt, A.; Rüterjans, H.; Chen, J.C.-H. Binding of a Designed Substrate Analogue to Diisopropyl Fluorophosphatase: Implications for the Phosphotriesterase Mechanism. J. Am. Chem. Soc. 2006, 128, 12750–12757, doi:10.1021/ja061887n.
- Soares, F.V.; De Castro, A.A.; Pereira, A.F.; Leal, D.H.S.; Mancini, D.T.; Krejcar, O.; Kuca, K.; Da Cunha, E.F.; Kuca, K. Theoretical Studies Applied to the Evaluation of the DFPase Bioremediation Potential against Chemical Warfare Agents Intoxication. Int. J. Mol. Sci. 2018, 19, 1257, doi:10.3390/ijms19041257.
- Wymore, T.; Field, M.J.; Langan, P.; Smith, J.C.; Parks, J.M. Hydrolysis of DFP and the Nerve Agent (S)-Sarin by DFPase Proceeds along Two Different Reaction Pathways: Implications for Engineering Bioscavengers. J. Phys. Chem. B 2014, 118, 4479–4489, doi:10.1021/jp410422c.
- Blum, M.-M.; Mustyakimov, M.; Rüterjans, H.; Kehe, K.; Schoenborn, B.P.; Langan, P.; Chen, J.C.-H. Rapid determination of hydrogen positions and protonation states of diisopropyl fluorophosphatase by joint neutron and X-ray diffraction refinement. Proc. Natl. Acad. Sci. USA 2009, 106, 713–718, doi:10.1073/pnas.0807842106.
- Hoskin, F.C.; Prusch, R.D. Characterization of a DFP-hydrolyzing enzyme in squid posterior salivary gland by use of soman, DFP and manganous ion. Comp. Biochem. Physiol. Part C Comp. Pharmacol. 1983, 75, 17–20, doi:10.1016/0742-8413(83)90004-x.
- Blum, M.-M.; Timperley, C.M.; Williams, G.R.; Thiermann, H.; Worek, F. Inhibitory Potency against Human Acetylcholinesterase and Enzymatic Hydrolysis of Fluorogenic Nerve Agent Mimics by Human Paraoxonase 1 and Squid Diisopropyl Fluorophosphatase. Biochemistry 2008, 47, 5216–5224, doi:10.1021/bi702222x.
- DeFrank, J.J.; Cheng, T.C. Purification and properties of an organophosphorus acid anhydrase from a halophilic bacterial isolate. J. Bacteriol. 1991, 173, 1938–1943, doi:10.1128/jb.173.6.1938-1943.1991.
- DeFrank, J.J.; Beaudry, W.T.; Cheng, T.-C.; Harvey, S.P.; Stroup, A.N.; Szafraniec, L.L. Screening of halophilic bacteria and Alteromonas species for organophosphorus hydrolyzing enzyme activity. Chem. Interact. 1993, 87, 141–148, doi:10.1016/0009-2797(93)90035-w.
- Cheng, T.-C.; Liu, L.; Wang, B.; Wu, J.; DeFrank, J.J.; Anderson, D.M.; Rastogi, V.K.; Hamilton, A.B. Nucleotide sequence of a gene encoding an organophosphorus nerve agent degrading enzyme from Alteromonas haloplanktis. J. Ind. Microbiol. Biotechnol. 1997, 18, 49–55, doi:10.1038/sj.jim.2900358.
- Cheng, T.-C.; Harvey, S.P.; Stroup, A.N. Purification and Properties of a Highly Active Organophosphorus Acid Anhydrolase from Alteromonas undina. Appl. Environ. Microbiol. 1993, 59, 3138–3140, doi:10.1128/aem.59.9.3138-3140.1993.
- Cheng, T.-C.; DeFrank, J.J. Hydrolysis of Organophosphorus Compounds by Bacterial Prolidases. Enzymes Action 2000, 243–261, doi:10.1007/978-94-010-0924-9_12.
- Vyas, N.K.; Nickitenko, A.; Rastogi, V.K.; Shah, S.S.; Quiocho, F.A. Structural Insights into the Dual Activities of the Nerve Agent Degrading Organophosphate Anhydrolase/Prolidase. Biochemistry 2010, 49, 547–559, doi:10.1021/bi9011989.
- Cheng, T.C.; DeFrank, J.J.; Rastogi, V.K. Alteromonas prolidase for organophosphorus G-agent decontamination. Chem. Interact. 1999, 119, 455–462, doi:10.1016/s0009-2797(99)00058-7.
- Cheng, T.C.; Harvey, S.P.; Chen, G.L. Cloning and expression of a gene encoding a bacterial enzyme for decontamination of organophosphorus nerve agents and nucleotide sequence of the enzyme. Appl. Environ. Microbiol. 1996, 62, 1636–1641, doi:10.1128/aem.62.5.1636-1641.1996.
- Daczkowski, C.M.; Pegan, S.D.; Harvey, S.P. Engineering the Organophosphorus Acid Anhydrolase Enzyme for Increased Catalytic Efficiency and Broadened Stereospecificity on Russian VX. Biochemistry 2015, 54, 6423–6433, doi:10.1021/acs.biochem.5b00624.
- Kitchener, R.; Grunden, A. Prolidase function in proline metabolism and its medical and biotechnological applications. J. Appl. Microbiol. 2012, 113, 233–247, doi:10.1111/j.1365-2672.2012.05310.x.
- Hill, C.M.; Wu, F.; Cheng, T.-C.; DeFrank, J.J.; Raushel, F.M. Substrate and stereochemical specificity of the organophosphorus acid anhydrolase from Alteromonas sp. JD6.5 toward p -nitrophenyl phosphotriesters. Bioorganic Med. Chem. Lett. 2000, 10, 1285–1288, doi:10.1016/s0960-894x(00)00213-4.


