 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Camila Xu | -- | 3462 | 2022-11-09 01:47:05 |
Video Upload Options
Non-nucleoside reverse-transcriptase inhibitors (NNRTIs) are antiretroviral drugs used in the treatment of human immunodeficiency virus (HIV). NNRTIs inhibit reverse transcriptase (RT), an enzyme that controls the replication of the genetic material of HIV. RT is one of the most popular targets in the field of antiretroviral drug development. Discovery and development of NNRTIs began in the late 1980s and in the end of 2009 four NNRTI had been approved by regulatory authorities and several others were undergoing clinical development. Drug resistance develops quickly if NNRTIs are administered as monotherapy and therefore NNRTIs are always given as part of combination therapy, the highly active antiretroviral therapy (HAART).
1. History
Acquired immunodeficiency syndrome (AIDS) is a leading cause of death in the world.[1] It was identified as a disease in 1981. Two years later the etiology agent for AIDS, the HIV was described.[2] HIV is a retrovirus and has two major serotypes, HIV-1 and HIV-2. The pandemic mostly involves HIV-1 while HIV-2 has lower morbidity rate and is mainly restricted to western Africa.[3]
In the year 2009 over 40 million people were infected worldwide with HIV and the number keeps on growing.[4] The vast majority of infected individuals live in the developing countries.[5]
HIV drugs do not cure HIV infection, but the treatment aims at improving the quality of patients´ lives and decreased mortality. [6]
25 antiretroviral drugs were available in 2009 for the treatment of HIV infection. The drugs belong to six different classes that act at different targets. The most popular target in the field of antiretroviral drug development is the HIV-1 reverse transcriptase (RT) enzyme.[1] There are two classes of drugs that target the HIV-1 RT enzyme, nucleoside/nucleotide reverse-transcriptase inhibitors (NRTIs/NtRTIs) and non-nucleoside reverse-transcriptase inhibitors (NNRTIs). Drugs in these classes are important components of the HIV combination therapy called highly active antiretroviral therapy, better known as HAART.[7]
In 1987, the first drug for the treatment of HIV infection was approved by the U.S. Food and Drug Administration (FDA). This was the NRTI called zidovudine. In the late 1980s, during further development of NRTIs, the field of NNRTIs discovery began. The development of NNRTIs improved quickly into the 1990s and they soon became the third class of antiretroviral drugs, following the protease inhibitors.[5][6]
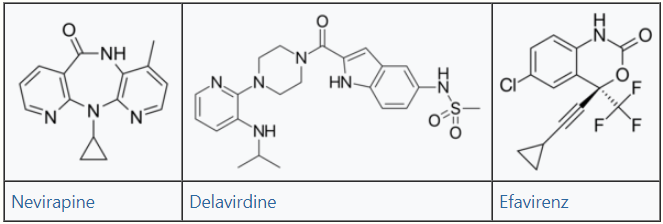
The NNRTIs are HIV-1 specific and have no activity against HIV-2 and other retroviruses. The first NNRTI, nevirapine was discovered by researchers at Boehringer Ingelheim and approved by the FDA in 1996. In the next two years two other NNRTIs were approved by the FDA, delavirdine in 1997 and efavirenz in 1998.[2] These three drugs are so-called first generation NNRTIs. The need for NNRTIs with better resistance profile led to the development of the next generation of NNRTIs. Researchers at Janssens Foundation and Tibotec discovered the first drug in this class, etravirine, which was approved by the FDA in 2008. The second drug in this class, rilpivirine, was also discovered by Tibotec and was approved by the FDA in 2011. In addition to these four NNRTIs several other are in clinical development.[5]
2. The HIV-1 Reverse Transcriptase Enzyme

2.1. Function
Reverse transcriptase (RT) is an enzyme that controls the replication of the genetic material of HIV and other retroviruses.[1] The enzyme has two enzymatic functions. Firstly it acts as a polymerase where it transcribes the single-stranded RNA genome into single-stranded DNA and subsequently builds a complementary strand of DNA. This provides a DNA double helix which can be integrated in the host cell's chromosome.[5] Secondly it has ribonuclease H (Rnase H) activity as it degrades the RNA strand of RNA-DNA intermediate that forms during viral DNA synthesis.[8]
2.2. Structure
The HIV-1 RT is an asymmetric 1000-amino acid heterodimer composed of p66 (560 amino acids) and p51 subunits (440 amino acids).[1] The p66 subunit has two domains, a polymerase and ribonuclease H. The polymerase domain contains four subdomains, which have been termed “fingers”, “palm”, “thumb” and “connection” and it is often compared to a right hand (figure 1).[5] The role of the p66 subunit is to carry out the activity of RT whereas it contains the active sites of the enzyme. The p51 is believed to play mainly a structural role.[8]
3. Binding and Pharmacophore
Despite the chemical diversity of NNRTIs they all bind at the same site in the RT. The binding occurs allosterically in a hydrophobic pocket located approximately 10 Å from the catalytic site in the palm domain of the p66 subunit site of the enzyme.[1][5] The NNRTI binding pocket (NNIBP) contains five aromatic (Tyr-181, Tyr-188, Phe-227 and Trp-229), six hydrophobic (Pro-59, Leu-100, Val-106, Val-179, Leu-234 and Pro-236) and five hydrophilic (Lys-101, Lys-103, Ser-105, Asp-132 and Glu-224) amino acids that belong to the p66 subunit and additional two amino acids (Ile-135 and Glu-138) belonging to the p51 subunit.[5] Each NNRTI interacts with different amino acid residues in the NNIBP.[9]

An important factor in the binding of the first generation NNRTIs, such as nevirapine, is the butterfly-like shape. Despite their chemical diversity they assume very similar butterfly-like shape.[9] Two aromatic rings of NNRTIs conform within the enzyme to resemble the wings of a butterfly (figure 2). The butterfly structure has a hydrophilic centre as a ‘body’ and two hydrophobic moieties representing the wings.[10] Wing I is usually a heteroaromatic ring and wing II is a phenyl or allyl substituent. Wing I has a functional group at one side of the ring which is capable of accepting and/or donating hydrogen bonds with the main chain of the amino acids Lys-101 and Lys-103. Wing II interacts through π-π interactions with a hydrophobic pocket, formed in most part by the side chains of aromatic amino acids. On the butterfly body a hydrophobic part fills a small pocket which is mainly formed by the side chains of Lys-103, Val-106 and Val-179.[11] However many other NNRTIs have been found to bind to RT in different modes. Second generation NNRTIs such as diarylpyrimidins (DAPYs), have a horseshoe-like shape with two lateral hydrophobic wings and a pyrimidine ring which is the central polar part.[12]
The NNIBP is elastic and the conformation depends on the size, specific chemical composition and binding mode of the NNRTI. The total structure of RT has segmental flexibility that depends on the nature of the bound NNRTI. It's important for the inhibitor to have flexibility to be able to bind in the modified pockets of a mutant target. Inhibitor flexibility may not affect the inhibitor-target interactions.[9]
4. Mechanism of Action
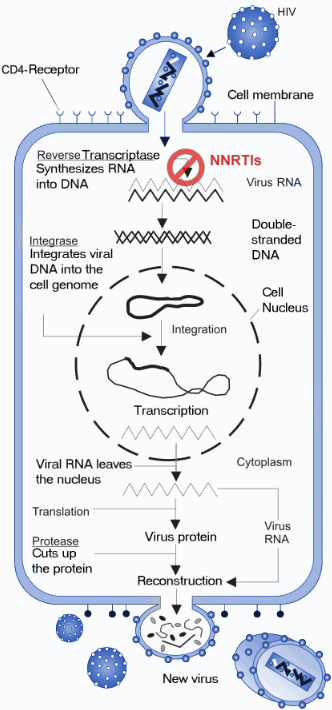
The NNRTIs act by binding non-competitively to the RT enzyme (figure 3). The binding causes conformational change in the three-dimensional structure of the enzyme and creates the NNIBP.[4][5] Binding of NNRTI to HIV-1 RT makes the p66 thumb domain hyper extended because it induces rotamer conformation changes in amino acid residues Tyr-181 and Tyr-188.[13] This affects the catalytic activity of the enzyme and blocks the HIV-1 replication by inhibiting the polymerase active site of the RT's p66 subunit.[14] The global conformational change additionally destabilizes the enzyme on its nucleic acid template and reduces its ability to bind nucleotides.[15] The transcription of the viral RNA is inhibited and therefore the replication rate of the virus reduces.[4] Although the exact molecular mechanism is still hypothetical this has been demonstrated by multiple studies to be the primary mechanism of action.
In addition to this proposed primary mechanism of action it has been shown that the NNRTIs have other mechanisms of action and interfere with various steps in the reverse transcriptase reaction.[5] It has been suggested that the inhibition of reverse transcription by the NNRTIs may be due to effects on the RT Rnase H activity and/or template/primer binding. Some NNRTIs interfere with HIV-1 Gag-Pol polyprotein processing by inhibiting the late stage of HIV-1 replication.
It is important to gain profound understanding of the various mechanism of action of the NNRTIs in order to develop next-generation NNRTIs and for understanding the mechanism of drug resistance.[14]
5. Drug Discovery and Design
The development of effective anti-HIV drugs is difficult due to wide variations in nucleotide and amino acid sequences. The perfect anti-HIV drug chemical should be effective against drug resistance mutation. Understanding the target RT enzyme and its structure, mechanism of drug action and the consequence of drug resistance mutations provide useful information which can be helpful to design more effective NNRTIs. The RT enzyme can undergo change due to mutations that can disturb NNRTI binding.[9]
5.1. Discovery
The first two classes of compounds that were identified as NNRTIs were the 1-(2-2-hydroxyethoxymethyl)-6-(phenylthio)thymine (HEPT) and tetrahydroimidazo[4,5,1-jkj][1,4]benzodiazepin-2(1H)-one and -thione (TIBO) compounds. The discovery of the TIBO compounds led to the definition of the NNRTI class in the late 1980s[16] when they were unexpectedly found to inhibit RT. This finding initiated researches on mechanism of action for these compounds. The HEPT compounds were described before the TIBO compounds and were originally believed to be NRTIs. Later it was discovered that they shared common mechanism of action with the TIBO compounds.[2][5] Both the HEPT and TIBO compounds were first to be identified as highly specific and potent HIV-1 RT inhibitors, not active against other RTs.[2] These compounds do not interrupt the cellular or mitochondrial DNA synthesis. The specificity of the NNRTIs for HIV-1 is considered the hallmark of the NNRTI drug class.[5]
5.2. Development
First generation NNRTIs
After the discovery of HEPT and TIBO, compounds screening methods were used to develop BI-RG-587, the first NNRTI commonly known as nevirapine. Like HEPT and TIBO, nevirapine blocked viral RT activity by non-competitive inhibition (with respect to dNTP binding). This reinforced the idea that the new class of anti-HIV inhibitors was inhibiting the activity of RT but not at the active site. Several molecular families of NNRTIs have emerged following screening and evolution of many molecules.[6]
Three NNRTI compounds of the first generation have been approved by the FDA for treating HIV-1 infection. Nevirapine was approved in 1996, delavirdine in 1997 and efavirenz in 1998 (table 1). Two of these drugs, nevirapine and efavirenz, are cornerstones of first line HAART while delavirdine is hardly used nowadays.[5][6] The structure of these three drugs show the wide array of rings, substituents, and bonds that allow activity against HIV-1 RT. This diversity demonstrates why so many non-nucleosides have been synthesised but doesn't explain why only three drugs have reached the market. The main problem has been the potency of these compounds to develop resistance.[6]
Table 1 First generation NNRTIs approved by the FDA.
Development from α-APA to ITU
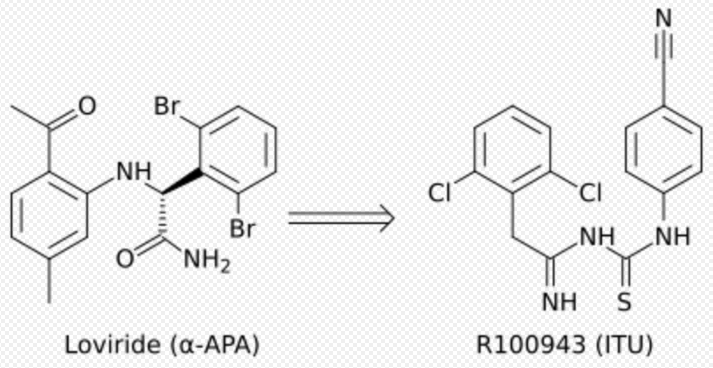
Crystal structure analysis showed that the first generation NNRTIs (for example TIBO, nevirapine and α-APA) bind HIV-1 RT in a “butterfly-like” conformation. These first generation NNRTIs were vulnerable against the common drug-resistance mutations like Tyr-181C and Tyr-188L/H. This triggered the need for finding new and more effective NNRTIs. ITU (imidoylthiourea), a promising series of NNRTIs emerged from α-APA analogs (figure 4). The ITU compounds were obtained by extending the linker that binds the aryl side groups of the α-APA. A potent ITU compound, R100943, was obtained by an arrangement of the chemical composition of the side groups based on structure-activity relationships (SAR). A crystal structure of the HIV-1/R100943 complex demonstrated that ITU compounds are more flexible than α-APA compound. The ITU compounds showed distinct mode of binding where they bound with "horseshoe" or "U" mode. The 2,6-dichlorophenyl part of R100943 which corresponds chemically to the wing II 2,6-dibromophenyl part of the α-APA occupied the wing I part in the NNIBP whereas the 4-cyanoanilino part of R100943 occupies the wing II position in the NNIBP.[9][17]
R100943 inhibited HIV-1 and was considerably effective against a number of key NNRTI-resistant mutants like G190A mutation, which caused high-level resistance to loviride (α-APA) and nevirapine. G190A mutation was thought to cause resistance by occupying a part of the binding pocket that would otherwise be filled by the linker part of the butterfly shaped NNRTIs. R100943, in the horseshoe mode of binding, is located at a distance of approximately 6.0 Å from G190. When compared with nevirapine and loviride which bind in the butterfly shape the ITU derivatives revealed improved activity against Tyr-181C and Tyr-188L mutants. A structural study suggested that a potent TIBO compound could partly supplement for the effects of the Tyr-181C mutation by moving itself in the non-nucleoside inhibitor binding pocket (NNIBP) of the mutant RT. In this context, R100943 has torsional freedom that enables the conformational alternations of the NNRTI. This torsional freedom could be used by the ITU derivate to bind to a mutated NNIBP and thus compensating for the effects of a resistance mutation. Nevertheless, the potency of R100943 against HIV-1 resistant mutants was not adequate for it to be considered as an effective drug candidate. Additionally, the chemical stability of the imidoylthiourea part of the ITU derivative was not favorable for an oral drug.[9][18][19]
Development from ITU to DATA

Changes in the imidoylthiourea complexes led to the synthesis of a new class of compounds, diaryltriazine (DATA). In these compounds, the thiourea part of the ITU compounds was replaced by a triazine ring. The DATA compounds were more potent than the ITU compounds against common NNRTI resistant mutant strains. R106168, a prototype DATA compound, was rather easy to synthesize. Multiple substitutions were made at different positions on all of the three rings and on the linkers connecting the rings. In the pocket, most of the DATA derivatives conformed a horseshoe conformation. The two wings in R106168 (2,6-dichlorobenzyl and 4-cyanoanilino) occupied positions in the pocket similar to that of the two wings of the derivatives of ITU. The central part of the DATA compounds, in which the triazine ring replaced the thiourea group of ITU derivatives, is positioned between the side chains of L100 and V179. This removed a number of torstional degrees of freedom in the central part while keeping the flexibility between the triazine ring and the wings.[9][20]
Chemical substitution or modification in the three-aromatic-ring backbone of the DATA compounds had substantial effect on the activity. R120393, a DATA analog, was designed with a chloroindole part in wing I to expand interactions with the side chain of conserved W229 of the polymerase primer grip loop. R120393 had similar effect as R106168 against most of the NNRTI-resistant mutants. The cloroindole part interacted with the hydrophobic core of the pocket and influenced the binding mode of the R120393 so it went deeper into the pocket compared to the wing I position of other DATA analogs. Crystal structures showed that the DATA compounds could bind the NNIBP in different conformations. The capability to bind in multible modes made the NNRTIs stronger against drug-resistance mutations. Variability between the inhibitors could be seen when the chemical composition, size of wing I and the two linker groups connecting the rings were altered. The potency of the NNRTIs changed when the triazine nitrogen atoms were substituted with carbons.[9]
Next generation NNRTIs
Researchers used multi-disciplinary approach to design NNRTIs with better resistance profile and an increased genetic barrier to the development of resistance.[9] A new class of compounds, diarylpyrimide (DAPY), were discovered with the replacement of the central triazine ring from the DATA compounds, with a pyrimidine. This new class was more effective against drug resistant HIV-1 strains than the corresponding DATA analogs. The replacement enabled substitutions to the CH-group at the 5-position of the central aromatic ring. One of the first DAPY compounds, dapivirine (with R1= 2,4,6-trimethylanilino, R2 = R3 = H and Y = NH) was found to be effective against drug-resistant HIV-1 strains. Systematic chemical substitutions were made at the R1, R2, R3 and Y positions to find new DAPY derivatives. This led to the discovery of etravirine which has a bromine substitution at the 5-position (R3) of the pyrimidine ring (with R1 = 2,6-dimethyl-4-cyanoanilino, R2 = NH2 and Y = O) (figure 5).[9] Etravirine was discovered by researchers at the Jansen Research Foundation and Tibotec and approved in 2008 by the FDA. It is used in treatment-expirenced adult patients with HIV infection that is multidrug resistant in combination with other antiretroviral drugs.[21]
6. Resistance
When treating infection, whether bacterial or viral, there is always a risk of the infectious agent to develop drug resistance. The treatment of HIV infection is especially susceptible to drug resistance which is a serious clinical concern in the chemotherapeutic treatment of the infection. Drug resistant HIV-strains emerge if the virus is able to replicate in the presence of the antiretroviral drugs.[9]
NNRTI-resistant HIV-strains have the occurring mutations mainly in and around the NNIBP affecting the NNRTI binding directly by altering the size, shape and polarity on different areas of the pocket or by affecting, indirectly, the access to the pocket.[9] Those mutations are primarily noted in domains which span amino acids 98-108, 178-190 or 225-238 of the p66 subunit. The most frequent mutations observed in viruses isolated from patients who have been on a failing NNRTI containing chemotherapy are Lys-103N and Tyr-181C. NNRTI resistance has been linked to over 40 amino acid substitutions in vitro and in vivo.[5]
Antiretroviral drugs are never used in monotherapy due to rapid resistance development. The highly active antiretroviral therapy (HAART) was introduced in 1996.[4] The treatment regimen combines three drugs from at least two different classes of antiretroviral drugs.[5]
The advance of etravirine over other NNRTIs is that multiple mutations are required for the development of drug resistance. The drug has also shown activity against viruses with common NNRTI resistance associated mutations and cross-resistance mutations.[21]
7. Current Status
Five drugs in the class of NNRTIs have been approved by regulatory authorities. These are the first generation NNRTIs nevirapine, delavirdine and efavirenz and the next generation NNRTIs etravirine, and rilpivirine. Several other NNRTIs underwent clinical development but were discontinued due to unfavourable pharmacokinetic, efficacy and/or safety factors. Currently there are four other NNRTIs undergoing clinical development, IDX899, RDEA-428 and lersivirine (table 2).
7.1. Rilpivirine
Rilpivirine is a DAPY compound like etravirine and was discovered when further optimization within this family of NNRTIs was conducted. The resistance profile and the genetic barrier to the development of resistance is comparable to etravirine in vitro. The advantage of rilpivirine over etravirine is a better bioavailability and it is easier to formulate than etravirine. Etravirine has required extensive chemical formulation work due to poor solubility and bioavailability.[5] Rilpivirine was undergoing phase III clinical trials in the end of 2009.[22] Rilpivirine was approved by the FDA for HIV therapy in May 2011 under the brand name Edurant.[23] Edurant is approved for treatment-naive patients with a viral load of 100,000 copies/mL or less at therapy initiation.[24] Its recommended dosage is 25 mg orally once daily with a meal, in combination with other antiretrovirals.[25] It is contraindicated for use with proton pump inhibitors due to the increased gastric pH causing decreased rilpivirine plasma concentrations, potentially resulting in loss of virologic response and possible resistance.[25] A fixed-dose drug combining rilpivirine with emtricitabine and tenofovir disoproxil (TDF), was approved by the U.S. Food and Drug Administration in August 2011 under the brand name Complera.[26] A newer fixed-dose drug also combining rilpivirine with emtricitabine and tenofovir alafenamide (TAF) was approved in March 2016 under the brand name Odefsey.
7.2. RDEA806
In 2007 a new family of triazole NNRTIs was presented by researchers from the pharmaceutical company Ardea Biosciences. The selected candidate from the screening executed was RDEA806 belonging to the family of triazoles. It has similar resistance profile against selected NNRTI resistant HIV-1 strains to other next generation NNRTIs.[5] The candidate entered phase IIb clinical trials in the end of 2009,[27], but no further trial have been initiated. Ardea was sold to AstraZeneca in 2012.[28]
7.3. Fosdevirine (IDX899)
Fosdevirine (also known as IDX899 and GSK-2248761) is another next generation NNRTI developed by Idenix Pharmaceuticals and ViiV Healthcare. It belongs to the family of 3-phosphoindoles. In vitro studies have shown comparable resistance profile to that of the other next generation NNRTIs.[5] In November 2009 the candidate entered phase II clinical trials, but the trial and all further development was halted when 5 of 35 subjects receiving fosdevirine experienced delayed-onset seizures.[29]
7.4. Lersivirine (UK-453061)
Lersivirine belongs to the pyrazole family and is another next generation NNRTI in clinical trials developed by the pharmaceutical company ViiV Healthcare. The resistance profile is similar to that of other next generation NNRTIs. In the end of 2009 lersivirine was in phase IIb.[5] In February 2013, ViiV Healthcare announced a stop of the development program investigating lersivirine.[30]
Table 2 Represents the drug candidates that have been in clinical development

References
- "Elucidating the inhibition mechanism of HIV-1 non-nucleoside reverse transcriptase inhibitors through multicopy molecular dynamics simulations". Journal of Molecular Biology 388 (3): 644–58. May 2009. doi:10.1016/j.jmb.2009.03.037. PMID 19324058. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2744402
- "Anti-HIV drugs: 25 compounds approved within 25 years after the discovery of HIV". International Journal of Antimicrobial Agents 33 (4): 307–20. April 2009. doi:10.1016/j.ijantimicag.2008.10.010. PMID 19108994. https://dx.doi.org/10.1016%2Fj.ijantimicag.2008.10.010
- "Structure of HIV-2 reverse transcriptase at 2.35-A resolution and the mechanism of resistance to non-nucleoside inhibitors". Proceedings of the National Academy of Sciences of the United States of America 99 (22): 14410–5. October 2002. doi:10.1073/pnas.222366699. PMID 12386343. Bibcode: 2002PNAS...9914410R. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=137897
- "Drug delivery systems in HIV pharmacotherapy: what has been done and the challenges standing ahead". Journal of Controlled Release 138 (1): 2–15. August 2009. doi:10.1016/j.jconrel.2009.05.007. PMID 19445981. https://dx.doi.org/10.1016%2Fj.jconrel.2009.05.007
- "Non-nucleoside reverse transcriptase inhibitors (NNRTIs), their discovery, development, and use in the treatment of HIV-1 infection: a review of the last 20 years (1989-2009)". Antiviral Research 85 (1): 75–90. January 2010. doi:10.1016/j.antiviral.2009.09.008. PMID 19781578. https://dx.doi.org/10.1016%2Fj.antiviral.2009.09.008
- "Reverse transcription of the HIV-1 pandemic". FASEB Journal 21 (14): 3795–808. December 2007. doi:10.1096/fj.07-8697rev. PMID 17639073. https://dx.doi.org/10.1096%2Ffj.07-8697rev
- "Novel HIV-1 reverse transcriptase inhibitors". Virus Research 134 (1–2): 171–85. June 2008. doi:10.1016/j.virusres.2008.01.003. PMID 18308412. https://dx.doi.org/10.1016%2Fj.virusres.2008.01.003
- "The search for potent, small molecule NNRTIs: A review". Bioorganic & Medicinal Chemistry 17 (16): 5744–62. August 2009. doi:10.1016/j.bmc.2009.06.060. PMID 19632850. https://dx.doi.org/10.1016%2Fj.bmc.2009.06.060
- "Crystallography and the design of anti-AIDS drugs: conformational flexibility and positional adaptability are important in the design of non-nucleoside HIV-1 reverse transcriptase inhibitors". Progress in Biophysics and Molecular Biology 88 (2): 209–31. June 2005. doi:10.1016/j.pbiomolbio.2004.07.001. PMID 15572156. https://dx.doi.org/10.1016%2Fj.pbiomolbio.2004.07.001
- "Synthesis and evaluation of anti-HIV activity of isatin beta-thiosemicarbazone derivatives". Bioorganic & Medicinal Chemistry Letters 15 (20): 4451–5. October 2005. doi:10.1016/j.bmcl.2005.07.046. PMID 16115762. https://dx.doi.org/10.1016%2Fj.bmcl.2005.07.046
- "Non-nucleoside HIV-1 reverse transcriptase inhibitors di-halo-indolyl aryl sulfones achieve tight binding to drug-resistant mutants by targeting the enzyme-substrate complex". Antiviral Research 81 (1): 47–55. January 2009. doi:10.1016/j.antiviral.2008.09.008. PMID 18984007. https://dx.doi.org/10.1016%2Fj.antiviral.2008.09.008
- "Powder for reconstitution of the anti-HIV-1 drug TMC278 - Formulation development, stability and animal studies". European Journal of Pharmaceutics and Biopharmaceutics 70 (3): 853–60. November 2008. doi:10.1016/j.ejpb.2008.06.030. PMID 18657611. https://dx.doi.org/10.1016%2Fj.ejpb.2008.06.030
- "Dawn of non-nucleoside inhibitor-based anti-HIV microbicides". The Journal of Antimicrobial Chemotherapy 57 (3): 411–23. March 2006. doi:10.1093/jac/dki464. PMID 16431862. https://dx.doi.org/10.1093%2Fjac%2Fdki464
- "Mechanisms of inhibition of HIV replication by non-nucleoside reverse transcriptase inhibitors". Virus Research 134 (1–2): 147–56. June 2008. doi:10.1016/j.virusres.2008.01.002. PMID 18372072. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2745993
- "Mechanism of allosteric inhibition of HIV-1 reverse transcriptase revealed by single-molecule and ensemble fluorescence". Nucleic Acids Research 42 (18): 11687–96. October 2014. doi:10.1093/nar/gku819. PMID 25232099. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4191400
- "Antiviral drug discovery and development: where chemistry meets with biomedicine". Antiviral Research 67 (2): 56–75. August 2005. doi:10.1016/j.antiviral.2005.05.001. PMID 16046240. https://dx.doi.org/10.1016%2Fj.antiviral.2005.05.001
- "Evolution of anti-HIV drug candidates. Part 1: From alpha-anilinophenylacetamide (alpha-APA) to imidoyl thiourea (ITU)". Bioorganic & Medicinal Chemistry Letters 11 (17): 2225–8. September 2001. doi:10.1016/S0960-894X(01)00410-3. PMID 11527703. https://dx.doi.org/10.1016%2FS0960-894X%2801%2900410-3
- "Structures of Tyr188Leu mutant and wild-type HIV-1 reverse transcriptase complexed with the non-nucleoside inhibitor HBY 097: inhibitor flexibility is a useful design feature for reducing drug resistance". Journal of Molecular Biology 284 (2): 313–23. November 1998. doi:10.1006/jmbi.1998.2171. PMID 9813120. https://dx.doi.org/10.1006%2Fjmbi.1998.2171
- "Crystal structures of 8-Cl and 9-Cl TIBO complexed with wild-type HIV-1 RT and 8-Cl TIBO complexed with the Tyr181Cys HIV-1 RT drug-resistant mutant". Journal of Molecular Biology 264 (5): 1085–100. December 1996. doi:10.1006/jmbi.1996.0698. PMID 9000632. https://dx.doi.org/10.1006%2Fjmbi.1996.0698
- "Evolution of anti-HIV drug candidates. Part 2: Diaryltriazine (DATA) analogues". Bioorganic & Medicinal Chemistry Letters 11 (17): 2229–34. September 2001. doi:10.1016/S0960-894X(01)00411-5. PMID 11527704. https://dx.doi.org/10.1016%2FS0960-894X%2801%2900411-5
- "Etravirine: a second-generation nonnucleoside reverse transcriptase inhibitor (NNRTI) active against NNRTI-resistant strains of HIV". Clinical Therapeutics 31 (4): 692–704. April 2009. doi:10.1016/j.clinthera.2009.04.020. PMID 19446143. https://dx.doi.org/10.1016%2Fj.clinthera.2009.04.020
- http://www.tibotec.com/bgdisplay.jhtml?itemname=HIV_tmc278
- "Approval of Edurant (rilpivirine), a new NNRTI, for the Treatment of HIV in Treatment Naive Patients" (Press release). Food and Drug Administration. May 20, 2011. Retrieved October 19, 2017. https://aidsinfo.nih.gov/news/902/approval-of-edurant--rilpivirine---a-new-nnrti--for-the-treatment-of-hiv-in-treatment-naive-patients---may-20--2011
- "[Data on rilpivirine in treatment-naïve patients. Lessons from ECHO, THRIVE and STaR]" (in Spanish). Enfermedades Infecciosas y Microbiologia Clinica 31 Suppl 2: 20–9. June 2013. doi:10.1016/S0213-005X(13)70139-3. PMID 24252530. https://dx.doi.org/10.1016%2FS0213-005X%2813%2970139-3
- https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/202022s000lbl.pdf
- "Approval of Complera: emtricitabine/rilpivirine/tenofovir DF fixed dose combination" (Press release). Food and Drug Administration. August 10, 2011. Retrieved October 19, 2017. https://aidsinfo.nih.gov/news/909/approval-of-complera--emtricitabine-rilpivirine-tenofovir-df-fixed-dose-combination---august-10--2011
- http://www.ardebio.com/ http://www.ardeabio.com/viral.html
- "AstraZeneca to acquire Ardea Biosciences for $1 billion (Net of existing cash) including lead product lesinurad in Phase III development for gout". https://www.astrazeneca.com/media-centre/press-releases/2012/AstraZeneca-to-acquire-Ardea-Biosciences-for-1-billion-23042012.html.
- [1]
- http://www.hivandhepatitis.com/ http://www.hivandhepatitis.com/hiv-aids/hiv-aids-topics/hiv-treatment/3963-viiv-announces-halt-to-lersivirine-development


