 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Camila Xu | -- | 2064 | 2022-09-30 01:37:12 |
Video Upload Options
The agouti gene encodes the agouti-signaling protein (ASIP), responsible for the distribution of melanin pigment in mammals. Agouti interacts with the melanocortin 1 receptor to determine whether the melanocyte (pigment cell) produces phaeomelanin (a red to yellow pigment), or eumelanin (a brown to black pigment). This interaction is responsible for making distinct light and dark bands in the hairs of animals such as the agouti. In other species such as horses, the agouti gene is responsible for determining which parts of the body will be black. Horses can either have black distributed across its body or can have black only in certain locations such as the tail, ears, mane, and the lower legs. In mice, the agouti gene determines whether the animal will be yellow or grey and brown. The agouti-signaling protein (ASIP) is a competitive antagonist with alpha-Melanocyte-stimulating hormone (α-MSH) to bind with melanocortin 1 receptor (MC1R) proteins. Activation by α-MSH causes production of the darker eumelanin, while activation by ASIP causes production of the redder phaeomelanin. This means that if agouti is expressed then the phenotype will be yellow and if it's not expressed then the phenotype would be grey.
1. Pigment Development
In mice, the wild type agouti allele (A) presents a grey phenotype, however, many allele variants have been identified through genetic analyses, which result in a wide range of phenotypes distinct from the typical grey coat.[1] The most widely studied allele variants are the lethal yellow mutation (Ay) and the viable yellow mutation (Avy) which are caused by ectopic expression of agouti.[1] These mutations are also associated with yellow obese syndrome which is characterized by early onset obesity, hyperinsulinemia and tumorigenesis.[1][2] The murine agouti gene locus is found on chromosome 2 and encodes a 131 amino acid protein. This protein signals the distribution of melanin pigments in epithelial melanocytes located at the base of hair follicles with expression being more sensitive on ventral hair than on dorsal hair.[3][4] Agouti is not directly secreted in the melanocyte as it works as a paracrine factor on dermal papillae cells to inhibit release of melanocortin.[5] Melanocortin acts on follicular melanocytes to increase production of eumelanin, a melanin pigment responsible for brown and black hair. When agouti is expressed, production of pheomelanin dominates, a melanin pigment that produces yellow or red colored hair.[6] Typically, it would be expected that the mice would be more brown and black due to evolution as this would be more advantageous in order to blend into its environment. Yet, mice are still born yellow due to the agouti gene allowing for pheomelanin expression instead of eumelanin expression.[3]
2. Mutations
The lethal yellow mutation (Ay) was the first embryonic mutation to be characterized in mice, as homozygous lethal yellow mice (Ay/ Ay) die early in development, due to an error in trophectoderm differentiation.[3] Lethal yellow homozygotes are rare today, while lethal yellow and viable yellow heterozygotes (Ay/a and Avy/a) remain more common. In wild-type mice agouti is only expressed in the skin during hair growth, but these dominant yellow mutations cause it to be expressed in other tissues as well.[7] This ectopic expression of the agouti gene is associated with the yellow obese syndrome, characterized by early onset obesity, hyperinsulinemia and tumorigenesis.[3]
The lethal yellow (Ay) mutation is due to an upstream deletion at the start site of agouti transcription. This deletion causes the genomic sequence of agouti to be lost, except the promoter and the first non-encoding exon of Raly, a ubiquitously expressed gene in mammals.[4] The coding exons of agouti are placed under the control of the Raly promoter, initiating ubiquitous expression of agouti, increasing production of pheomelanin over eumelanin and resulting in the development of a yellow phenotype.[8]
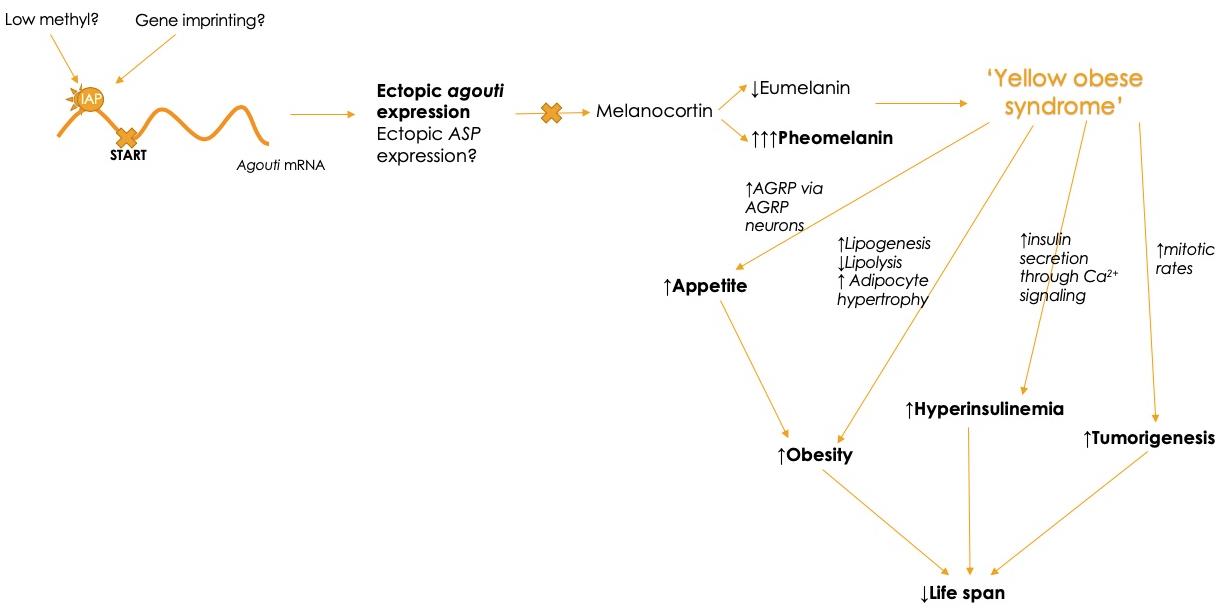
The viable yellow (Avy) mutation is due to a change in the mRNA length of agouti, as the expressed gene becomes longer than the normal gene length of agouti. This is caused by the insertion of a single intracisternal A particle (IAP) retrotransposon upstream to the start site of agouti transcription.[9] In the proximal end of the gene, an unknown promoter then causes agouti to be constitutionally activated, and individuals to present with phenotypes consistent with the lethal yellow mutation. Although the mechanism for the activation of the promoter controlling the viable yellow mutation is unknown, the strength of coat color has been correlated with the degree of gene methylation, which is determined by maternal diet and environmental exposure.[9] As agouti itself inhibits melanocortin receptors responsible for eumelanin production, the yellow phenotype is exacerbated in both lethal yellow and viable yellow mutations as agouti gene expression is increased. Agouti is unique because although it is a recessive allele, heterozygotes will appear yellow, not the dominant brown or black.[10]
Viable yellow (Avy/a) and lethal yellow (Ay/a) heterozygotes have shortened life spans and increased risks for developing early onset obesity, type II diabetes mellitus and various tumors.[5][11] The increased risk of developing obesity is due to the dysregulation of appetite, as agouti agonizes the agouti-related protein (AGRP), responsible for the stimulation of appetite via hypothalamic NPY/AGRP orexigenic neurons.[9] Agouti also promotes obesity by antagonizing melanocyte-stimulating hormone (MSH) at the melanocortin receptor (MC4R), as MC4R is responsible for regulating food intake by inhibiting appetite signals.[12] The increase in appetite is coupled to alterations in nutrient metabolism due to the paracrine actions of agouti on adipose tissue, increasing levels of hepatic lipogenesis, decreasing levels of lipolysis and increasing adipocyte hypertrophy.[13] This increases body mass and leads to difficulties with weight loss as metabolic pathways become dysregulated. Hyperinsulinemia is caused by mutations to agouti, as the agouti protein functions in a calcium dependent manner to increase insulin secretion in pancreatic beta cells, increasing risks of insulin resistance.[14] Increased tumor formation is due to the increased mitotic rates of agouti, which are localized to epithelial and mesenchymal tissues.[8]
3. Methylation and Diet Intervention

Correct functioning of agouti requires DNA methylation. Methylation occurs in six guanine-cytosine (GC) rich sequences in the 5’ long terminal repeat of the IAP element in the viable yellow mutation.[11] Methylation on a gene causes the gene to not be expressed because it will cause the promoter to be turned off. In utero, the mother's diet can cause methylation or demethylation. When this area is unmethylated, ectopic expression of agouti occurs, and yellow phenotypes are shown because the phaeomelanin is expressed instead of eumelanin. When the region is methylated, agouti is expressed normally, and grey and brown phenotypes (eumelanin) occur. The epigenetic state of the IAP element is determined by the level of methylation, as individuals show a wide range of phenotypes based on their degree of DNA methylation.[11] Increased methylation is correlated with increased expression of the normal agouti gene. Low levels of methylation can induce gene imprinting which results in offspring displaying consistent phenotypes to their parents, as ectopic expression of agouti is inherited through non-genomic mechanisms.[9][15]
DNA methylation is determined in utero by maternal nutrition and environmental exposure.[11] Methyl is synthesized de novo but attained through the diet by folic acid, methionine, betaine, and choline, as these nutrients feed into a consistent metabolic pathway for methyl synthesis.[16] Adequate zinc and vitamin B12 are required for methyl synthesis as they act as cofactors for transferring methyl groups.[17]
When inadequate methyl is available during early embryonic development, DNA methylation cannot occur, which increases ectopic expression of agouti and results in the presentation of the lethal yellow and viable yellow phenotypes which persist into adulthood. This leads to the development of the yellow obese syndrome, which impairs normal development and increases susceptibility to the development of chronic disease. Ensuring maternal diets are high in methyl equivalents is a key preventive measure for reducing ectopic expression of agouti in offspring. Diet intervention through methyl supplementation reduces imprinting at the agouti locus, as increased methyl consumption causes the IAP element to become completely methylated and ectopic expression of agouti to be reduced.[18] This lowers the proportion of offspring that present with the yellow phenotype and increases the number offspring that resemble agouti wild type mice with grey coats.[9] Two genetically identical mice could look very different phenotypically due the mothers' diets while the mice were in utero. If the mice has the agouti gene it can be expressed due to the mother eating a typical diet and the offspring would have a yellow coat. If the same mother had eaten a methyl-rich diet supplemented with zinc, vitamin B12, and folic acid then the offspring's agouti gene would likely become methylated, it wouldn't be expressed, and the coat color would be brown instead. In mice, the yellow coat color is also associated with health problems in mice including obesity and diabetes.[19]
4. Human Homologue
Agouti signaling protein (ASP) is the human homologue of murine agouti. It is encoded by the human agouti gene on chromosome 20 and is a protein consisting of 132 amino acids. It is expressed much more broadly than murine agouti and is found in adipose tissue, pancreas, testes, and ovaries, whereas murine agouti is solely expressed in melanocytes.[17] ASP has 85% similarity to the murine form of agouti.[20] As ectopic expression of murine agouti leads to the development of the yellow obese syndrome, this is expected to be consistent in humans.[20] The yellow obese syndrome increases the development of many chronic diseases, including obesity, type II diabetes mellitus and tumorigenesis.[1]
ASP has similar pharmacological activation to murine agouti, as melanocortin receptors are inhibited through competitive antagonism.[21] Inhibition of melanocortin by ASP can also be through non-competitive methods, broadening its range of effects.[8] The function of ASP differs to murine agouti. ASP effects the quality of hair pigmentation whereas murine agouti controls the distribution of pigments that determine coat color.[9] ASP has neuroendocrine functions consistent with murine agouti, as it agonizes via AgRP neurons in the hypothalamus and antagonizes MSH at MC4Rs which reduce satiety signals. AgRP acts as an appetite stimulator and increases appetite while decreasing metabolism. Because of these mechanisms, AgRP may be linked to increased body mass and obesity in both humans and mice.[22] Over-expression of AgRP has been linked to obesity in males, while certain polymorphisms of AgRP have been linked to eating disorders like anorexia nervosa.[23][24] The mechanism underlying hyperinsulinemia in humans is consistent with murine agouti, as insulin secretion is heightened through calcium sensitive signaling in pancreatic beta cells.[17] The mechanism for ASP induced tumorigenesis remains unknown in humans.[17]
4.1. Human Implications
Since research has suggested associations in humans between obesity, eating disorders, and agouti-signaling protein it might be beneficial for women who are pregnant to ensure they are eating a well-balanced diet in order to possibly help to improve the epigenome (what genes are expressed) of their child. It was found that in mice who were fed methyl-rich foods and vitamins like zinc, B12, folic acid, methionine, betaine, and choline (all of which are required for methyl synthesis) their offspring were healthy and brown, indicating that the agouti gene had not been expressed.[16] In genetically identical mice whose mothers were not fed methyl-rich foods, agouti expression occurred and the offspring were overweight and yellow.[16] It has been known that we as individuals can affect our epigenome through our diet and other environmental factors, but recently it has been implicated that while in utero our epigenome can also be affected.[25] It had been the general consensus that while we could not change our own genome (as it was passed down from our parents), we could in theory positively or negatively affect our epigenome through actions such as smoking, drinking, and eating healthy.[25] Since methylation is one of the most common ways for DNA to be modified, resulting in changes to the epigenome, the diets of the pregnant mice having such a drastic phenotypic effect on the offspring may imply a similar mechanism or scenario in humans.[16] As further research is done, it may become more apparent that not only our genome but also our epigenome can be greatly impacted before we are even born.
References
- "Molecular characterization of the mouse agouti locus". Cell 71 (7): 1195–204. December 1992. doi:10.1016/S0092-8674(05)80067-4. PMID 1473152. https://dx.doi.org/10.1016%2FS0092-8674%2805%2980067-4
- "Physiological consequences of ectopic agouti gene expression: the yellow obese mouse syndrome". Physiological Genomics 1 (3): 151–63. November 1999. doi:10.1152/physiolgenomics.1999.1.3.151. PMID 11015573. https://semanticscholar.org/paper/9191ff6d62ae4e71d904b3e9f1c35b930eb02a5a.
- "Mesoderm-ectoderm interaction in the production of the agouti pigmentation pattern in mice". Genetics 71 (2): 297–303. June 1972. PMID 4558326. PMC 1212784. https://www.genetics.org/content/genetics/71/2/297.full.pdf.
- Melmed, S. (Ed) (2010). The Pituitary (3rd ed.). Cambridge: MA: Academic Press.
- "The role of the agouti gene in the yellow obese syndrome". The Journal of Nutrition 127 (9): 1902S–1907S. September 1997. doi:10.1093/jn/127.9.1902S. PMID 9278579. https://dx.doi.org/10.1093%2Fjn%2F127.9.1902S
- "Agouti protein is an antagonist of the melanocyte-stimulating-hormone receptor". Nature 371 (6500): 799–802. October 1994. doi:10.1038/371799a0. PMID 7935841. Bibcode: 1994Natur.371..799L. https://dx.doi.org/10.1038%2F371799a0
- "Ectopic expression of the agouti gene in transgenic mice causes obesity, features of type II diabetes, and yellow fur". Proceedings of the National Academy of Sciences of the United States of America 92 (11): 4728–32. May 1995. doi:10.1073/pnas.92.11.4728. PMID 7761391. Bibcode: 1995PNAS...92.4728K. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=41780
- Tollefsbol, T. (Ed.) (2012). Epigenetics in Human Disease (6 ed.). Cambridge: MA: Academic Press.
- "The agouti mouse model: an epigenetic biosensor for nutritional and environmental alterations on the fetal epigenome". Nutrition Reviews 66 Suppl 1 (1): S7-11. August 2008. doi:10.1111/j.1753-4887.2008.00056.x. PMID 18673496. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2822875
- Barsh, G. S. (2001-01-01), "Agouti", in Brenner, Sydney; Miller, Jefferey H., Academic Press, pp. 23, doi:10.1006/rwgn.2001.0017, ISBN 9780122270802, http://www.sciencedirect.com/science/article/pii/B0122270800000173, retrieved 2019-09-19
- "Adipogenesis and obesity: rounding out the big picture". Cell 87 (3): 377–89. November 1996. doi:10.1016/S0092-8674(00)81359-8. PMID 8898192. https://dx.doi.org/10.1016%2FS0092-8674%2800%2981359-8
- "The MC4 receptor and control of appetite". British Journal of Pharmacology 149 (7): 815–27. December 2006. doi:10.1038/sj.bjp.0706929. PMID 17043670. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2014686
- "Cellularity of adipose depots in six strains of genetically obese mice". Journal of Lipid Research 13 (1): 2–11. January 1972. PMID 5059196. http://www.jlr.org/content/13/1/2.full.pdf.
- "The yellow mouse obesity syndrome and mechanisms of agouti-induced obesity". Obesity Research 7 (5): 506–14. September 1999. doi:10.1002/j.1550-8528.1999.tb00440.x. PMID 10509609. https://dx.doi.org/10.1002%2Fj.1550-8528.1999.tb00440.x
- null
- "Maternal methyl supplements in mice affect epigenetic variation and DNA methylation of offspring". The Journal of Nutrition 132 (8 Suppl): 2393S–2400S. August 2002. doi:10.1093/jn/132.8.2393S. PMID 12163699. https://dx.doi.org/10.1093%2Fjn%2F132.8.2393S
- "Structure and function of ASP, the human homolog of the mouse agouti gene". Human Molecular Genetics 4 (2): 223–30. February 1995. doi:10.1093/hmg/4.2.223. PMID 7757071. https://dx.doi.org/10.1093%2Fhmg%2F4.2.223
- "The insulin-like growth factor pathway as a target for cancer therapy". Clinical & Translational Oncology 12 (5): 326–38. May 2010. doi:10.1007/s12094-010-0514-8. PMID 20466617. https://dx.doi.org/10.1007%2Fs12094-010-0514-8
- "Nutrition & the Epigenome". https://learn.genetics.utah.edu/content/epigenetics/nutrition/.
- "Molecular structure and chromosomal mapping of the human homolog of the agouti gene". Proceedings of the National Academy of Sciences of the United States of America 91 (21): 9760–4. October 1994. doi:10.1073/pnas.91.21.9760. PMID 7937887. Bibcode: 1994PNAS...91.9760K. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=44896
- Handbook of Hormones. Cambridge: MA: Academic Press. 2015. pp. 66–67.
- "Hypothalamic expression of ART, a novel gene related to agouti, is up-regulated in obese and diabetic mutant mice". Genes & Development 11 (5): 593–602. March 1997. doi:10.1101/gad.11.5.593. PMID 9119224. https://dx.doi.org/10.1101%2Fgad.11.5.593
- "Plasma levels of agouti-related protein are increased in obese men". The Journal of Clinical Endocrinology and Metabolism 86 (5): 1921–4. May 2001. doi:10.1210/jcem.86.5.7458. PMID 11344185. https://dx.doi.org/10.1210%2Fjcem.86.5.7458
- "Association between an agouti-related protein gene polymorphism and anorexia nervosa". Molecular Psychiatry 6 (3): 325–8. May 2001. doi:10.1038/sj.mp.4000854. PMID 11326303. https://dx.doi.org/10.1038%2Fsj.mp.4000854
- "Epigenomics Fact Sheet" (in en). https://www.genome.gov/about-genomics/fact-sheets/Epigenomics-Fact-Sheet.

