 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Rita Xu | -- | 2802 | 2022-11-08 01:34:41 |
Video Upload Options
A cannabinoid receptor antagonist, also known simply as a cannabinoid antagonist or as an anticannabinoid, is a type of cannabinoidergic drug that binds to cannabinoid receptors (CBR) and prevents their activation by endocannabinoids. They include antagonists, inverse agonists, and antibodies of CBRs. The discovery of the endocannabinoid system led to the development of CB1 receptor antagonists. The first CBR inverse agonist, rimonabant, was described in 1994. Rimonabant blocks the CB1 receptor selectively and has been shown to decrease food intake and regulate body-weight gain. The prevalence of obesity worldwide is increasing dramatically and has a great impact on public health. The lack of efficient and well-tolerated drugs to cure obesity has led to an increased interest in research and development of CBR antagonists. Cannabidiol (CBD), a naturally occurring cannabinoid, is a non-competitive CB1/CB2 receptor antagonist. And Δ9-tetrahydrocannabivarin (THCV), a naturally occurring cannabinoid, modulate the effects of THC via direct blockade of cannabinoid CB1 receptors, thus behaving like first-generation CB1 receptor inverse agonists, such as rimonabant. CBD is a very low-affinity CB1 ligand, that can nevertheless affect CB1 receptor activity in vivo in an indirect manner, while THCV is a high-affinity CB1 receptor ligand and potent antagonist in vitro and yet only occasionally produces effects in vivo resulting from CB1 receptor antagonism. THCV has also high affinity for CB2 receptors and signals as a partial agonist, differing from both CBD and rimonabant.
1. History

For centuries hashish and marijuana from the Indian hemp Cannabis sativa L. have been used for medicinal and recreational purposes.[1][2] In 1840, Schlesinger S. was apparently the first investigator to obtain an active extract from the leaves and flowers of hemp.[3] A few years later, in 1848, Decourtive E. described the preparation of an ethanol extract that on evaporation of the solvent gave a dark resin, which he named “cannabin”.[4][5] In 1964 the main active constituent of C. sativa L., Δ9-tetrahydrocannabinol (THC), was isolated and synthesized by Mechoulam's laboratory.[1][6] Two types of cannabinoid receptors, CB1 and CB2, responsible for the effects of THC were discovered and cloned in the early 1990s.[7][8] Once cannabinoid receptors had been discovered, it became important to establish whether their agonists occur naturally in the body. This search led to the discovery of the first endogenous cannabinoid (endocannabinoid), anandamide (arachidonoyl ethanolamide). Later on other endocannabinoids were found, for example 2-AG (2-arachidonoyl glycerol).[1] These findings raised further questions about the pharmacological and physiological role of the cannabinoid system. This revived the research on cannabinoid receptor antagonists which were expected to help answer these questions.[8] The use of the cannabinoid agonist, THC, in its many preparations to enhance appetite is a well known fact. This fact led to the logical extension that blocking of the cannabinoid receptors might be useful in decreasing appetite and food intake.[9] It was then discovered that the blockage of the CB1 receptor represented a new pharmacological target. The first specific CB1 receptor antagonist / inverse agonist was rimonabant, discovered in 1994.[8][9][10]
2. Endocannabinoids and Their Signaling System
The endogenous cannabinoid system includes cannabinoid receptors, their endogenous ligands (endocannabinoids) and enzymes for their synthesis and degradation.[11]
There are two main receptor types associated with the endocannabinoid signaling system: cannabinoid receptor 1 (CB1) and 2 (CB2). Both receptors are 7-transmembrane G-protein coupled receptors (GPCRs) which inhibit the accumulation of cyclic adenosine monophosphate within cells.[12][13] CB1 receptors are present in highest concentration in the brain but can also be found in the periphery. CB2 receptors are mostly located in the immune and haematopoietic systems.[7][12]
Endocannabinoids are eicosanoids acting as agonists for cannabinoid receptors, and they occur naturally in the body.[6] Cannabinoid receptor-related processes are, for example, involved in cognition; memory; anxiety; control of appetite; emesis; motor behavior; sensory, autonomic, neuroendocrine, and immune responses; and inflammatory effects.[11] There are two well-characterized endocannabinoids located in the brain and periphery. The first identified was anandamide (arachidonoyl ethanolamide), and the second was 2-AG (2-arachidonoyl glycerol). Additional endocannabinoids include virodhamine (O-arachidonoyl ethanolamine), noladin ether (2-arachidonoyl glyceryl ether) and NADA (N-arachidonoyl dopamine).[12]
3. Mechanism of Action
CB1 receptors are coupled through Gi/o proteins and inhibit adenylyl cyclase and activate mitogen-activated protein (MAP) kinase. In addition, CB1 receptors inhibit presynaptic N- and P/Q-type calcium channels and activate inwardly rectifying potassium channels.[1][9] CB1 antagonists produce inverse cannabimimetic effects that are opposite in direction from those produced by agonists for these receptors.[1][14]
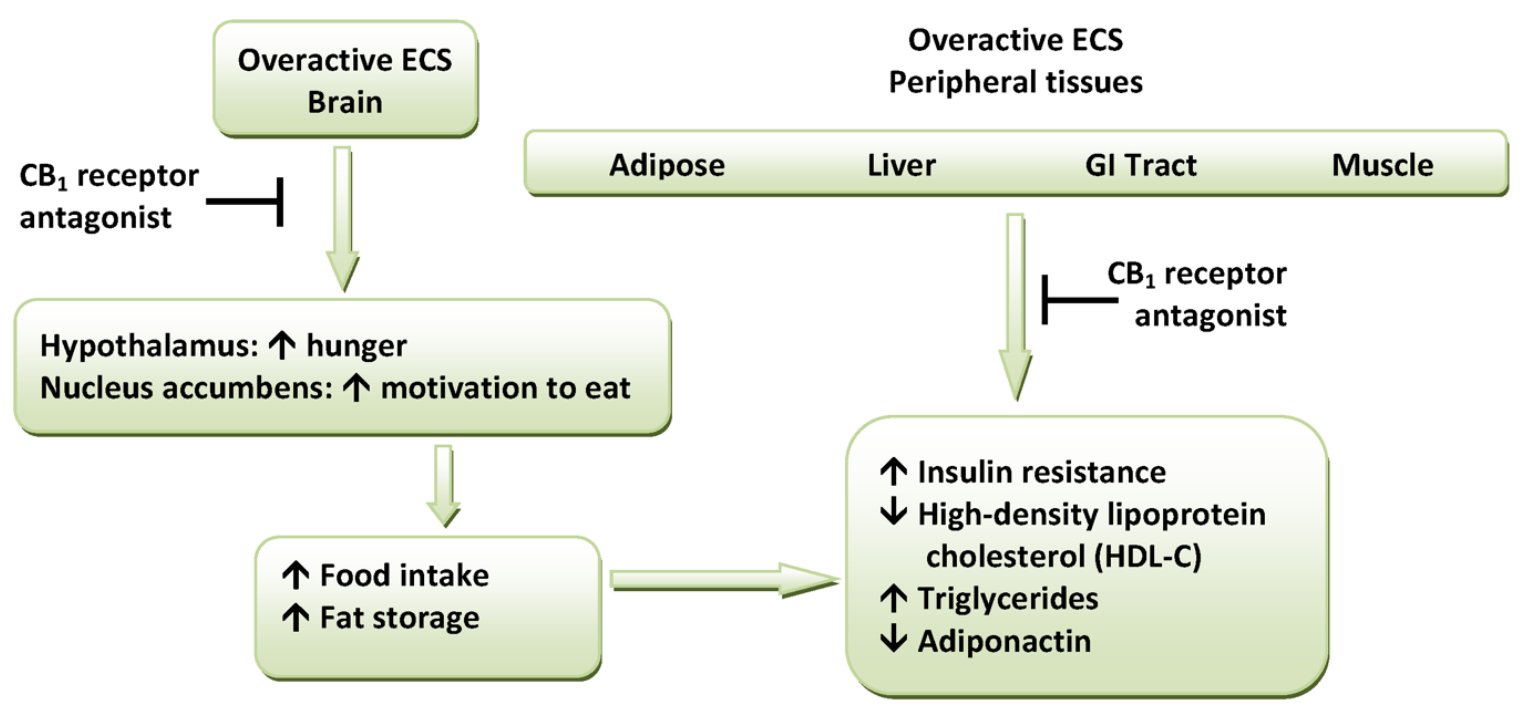
CB1 receptors are highly expressed in hypothalamic areas which are involved in central food intake control and feeding behavior. This strongly indicates that the cannabinoid system is directly involved in feeding regulation. These regions are also interconnected with the mesolimbic dopamine pathway, the so-called "reward" system. Therefore, CB1 antagonists might indirectly inhibit the dopamine-mediated rewarding properties of food.[12][14] Peripheral CB1 receptors are located in the gastrointestinal (GI) tract, liver and in adipose tissue. In the GI, CB1 receptors are located on nerve terminals in the intestines. Endocannabinoids act at the CB1 receptors to increase hunger and promote feeding and it is speculated that they decrease intestinal peristalsis and gastric emptying. Thus, antagonism at these receptors can inverse these effects.[12] Also, in peripheral tissues, antagonism of CB1 receptors increases insulin sensitivity and oxidation of fatty acids in muscles and the liver.[7] A hypothetical scheme for the metabolic effects of CB1 receptor antagonists is shown in Figure 1.
4. Drug Design
The first approach to develop cannabinoid antagonists in the late 1980s was to modify the structure of THC, but the results were disappointing. In the early 1990s new family of cannabinoid agonists was discovered from the NSAID (non-steroidal anti-inflammatory) drug pravadoline which led to the discovery of aminoalkyl indole antagonists with some but limited success. As the search based on the structure of agonists was disappointing it was no surprise that the first potent and selective cannabinoid antagonist belonged to an entirely new chemical family. In 1994 the first selective cannabinoid antagonist, SR141716 (rimonabant), was introduced by Sanofi belonging to a family of 1,5-diarylpyrazoles.[8][15]
4.1. Rimonabant
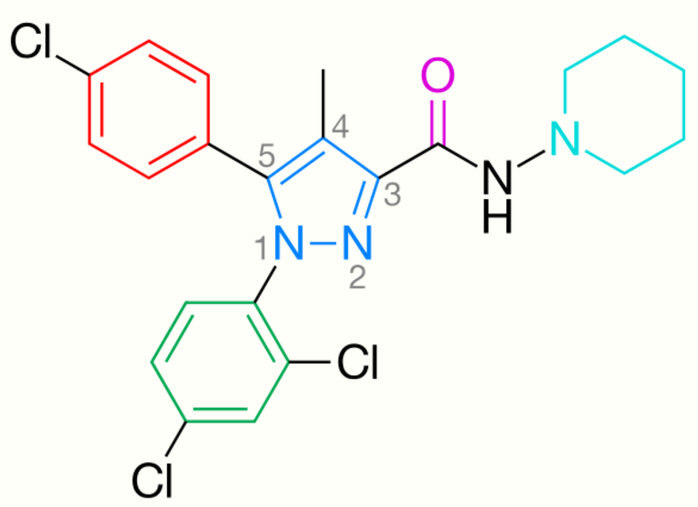
Rimonabant, also known by the systematic name [N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamidehydrochloride)], is a 1,5-diarylpyrazole CB1 receptor antagonist (Figure 2).[15] Rimonabant is not only a potent and highly selective ligand of the CB1 receptor, but it is also orally active and antagonizes most of the effects of cannabinoid agonists, such as THC, both in vitro and in vivo. Rimonabant has shown clear clinical efficacy for the treatment of obesity.[16]
4.2. Binding
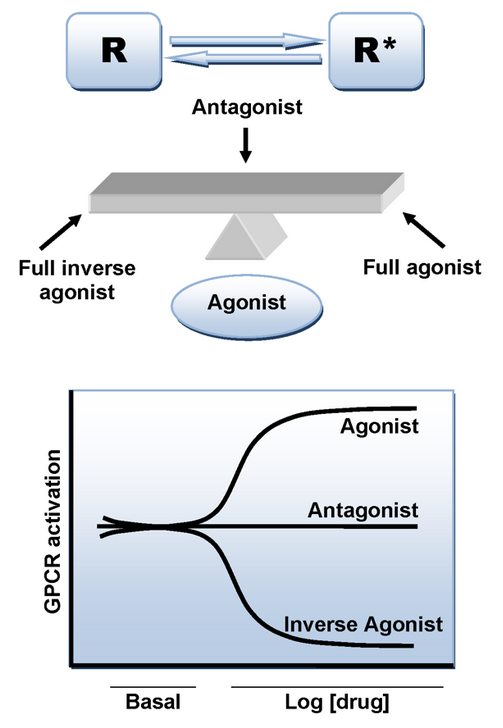
Binding of an agonist ligand to the CB1 receptor provokes a conformational change and leads to the active state of the receptor which is responsible for the signal transduction. However, there is an additional mechanism that can lead to the active state in the absence of ligand. As numerous other GPCRs, CB1 receptor displays a high level of constitutive activity and thus it can spontaneously adopt an active conformational state in the absence of agonist binding, keeping elevated basal levels of intracellular signaling.[17] This can be explained by the two state-model of receptor activation in which receptors are in equilibrium between two states, active and inactive (R* and R). An agonist will stabilize the active state leading to activation, a neutral antagonist binds equally to active and inactive states, whereas an inverse agonist will preferentially stabilize the inactive state (Figure 3).[17]
Rimonabant has been reported in many cases to behave as an inverse agonist rather than as a neutral antagonist and it is likely that it binds preferentially to the inactive state of the CB1, thereby decreasing the activation of the signaling pathway.[18][19] The key binding interaction is a hydrogen bond formed between the carbonyl group of rimonabant and the Lys192 residue of the CB1 receptor. This bond stabilizes the Lys192-Asp366 salt bridge of the intracellular end of transmembrane helices 3 and 6 (Figure 4). This specific salt bridge is present in the inactive state of the receptor but absent in the active state.[18][19]
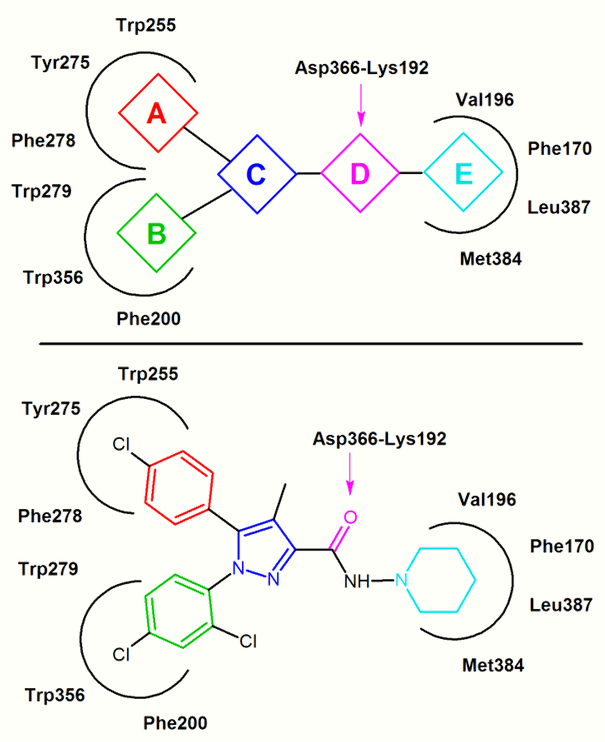
In the inactive state of CB1 rimonabant binds within the transmembrane-3-4-5-6 aromatic microdomain. The binding of rimonabant involves direct aromatic stacking interactions between its 2,4-dichlorophenyl ring and the Trp279/Phe200/Trp356 residues on the one side and the para-chlorophenyl ring and the Tyr275/Trp255/Phe278 residues on the other side. The lipophilic piperidinyl moiety fits nicely in a cavity formed by the amino acid residues Val196/Phe170/Leu387 and Met384 (Figure 4).[16][17][18][20]
4.3. Pharmacophore
Most CB1 antagonists reported so far are close analogs or isosteres of rimonabant.[21] A general CB1 inverse agonist pharmacophore model can be extracted from the common features of these analogs, diarylpyrazoles (Figure 4).[18] This pharmacophore contains a cyclic core, C, (e.g. pyrazole in rimonabant) substituted by two aromatic moieties, A and B. A hydrogen bond acceptor unit, D, connects C with a cyclic lipophilic part, E. In some cases unit E directly connects to C.[18][21] In Figure 4 rimonabant is used as an example. Unit A represents a 4-chlorophenyl group and unit B a 2,4-dichlorophenyl ring. Unit C is the central pyrazole ring and unit D represents the carbonyl group which serves as the hydrogen bond acceptor. Unit E represents a lipophilic aminopiperidinyl moiety.[18]
4.4. Structure-Activity Relationships
Optimal binding at the CB1 receptor requires a para-substituted phenyl ring at the pyrazole 5-position. The 5-substituent of the pyrazole is involved in receptor recognition and antagonism. The para-substituent of the phenyl ring could be chlorine, bromine or iodine, but it has been shown that an alkyl chain could also be tolerated.[18] Numbering of the central pyrazole ring is shown in Figure 2.
A 2,4-dichloro-substituted phenyl ring at the pyrazole 1-position is preferred for affinity as well as for the activity. It has been shown that additional halogens on this phenyl ring decrease affinity.[18]
It is also favorable to have a ring substitution at the 3-carboxamide group, such as the 1-piperidinyl group in rimonabant.[18] Replacement of the amino piperidinyl substituent by alkyl amides, ethers, ketones, alcohols or alkanes resulted mostly in decreased affinity. Replacement of the piperidinyl by pentyl or a heptyl chain gave the compounds agonistic properties. Based on these results it was concluded that the pyrazole 3-position seems to be involved in agonism, while the 1-,4-,5-positions appear to be involved in antagonism.[16]
Research has shown that the absence of the carboxamide oxygen results in decreased affinity. Furthermore, the presence of carboxamide oxygen contributes in conferring the inverse agonist properties, whereas analogs lacking this oxygen are found to be neutral antagonists. These results support the hypothesis that the carboxamide oxygen forms a hydrogen bond with Lys192 residue at the CB1 receptor.[22]
4.5. Diarylpyrazole Derivatives
SR141716 (rimonabant) analogs have recently been described by several groups, leading to a good understanding of the structure-activity relationship (SAR) within this chemical group. While most compounds described are less potent than SR141716, two of them are worth mentioning, SR147778 and AM251, although both may have action at mu opioid receptors as well.[23][24]
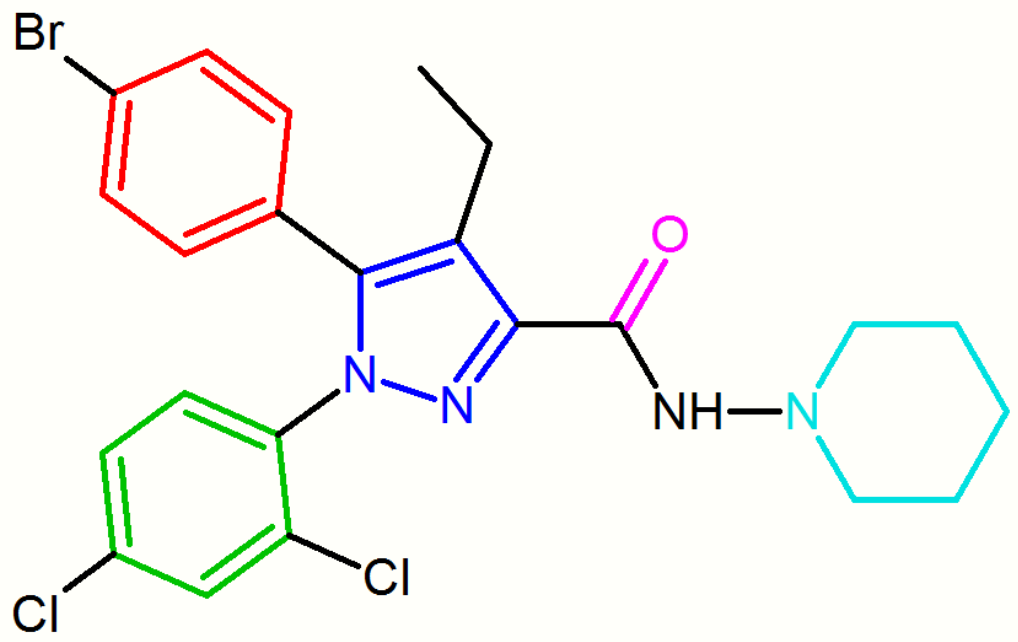
SR147778 (surinabant), a second generation antagonist, has a longer duration of action than rimonabant and enhanced oral activity. This enhanced duration of action is probably due to the presence of the more metabolically stable ethyl group at the 4-position of its pyrazole ring. Another change is the replacement of the 5-phenyl chlorine substituent by bromine.[18][24][25]
The diarylpyrazole derivative, AM251, has been described where chlorine substituent has been replaced by iodine in the para position of the 5-phenyl ring. This derivative appeared to be more potent and selective than rimonabant.[9][16]
21 analogs possessing either an alkyl amide or an alkyl hydrazide of variant lengths in position 3 were synthesized. It was observed that affinity increases with increased carbon chain length up to five carbons. Also the amide analogs exhibited higher affinity than hydrazide analogs. However, none of these analogs possessed significantly greater affinity than rimonabant but nevertheless, they were slightly more selective than rimonabant for the CB1 receptor over the CB2 receptor.[16]
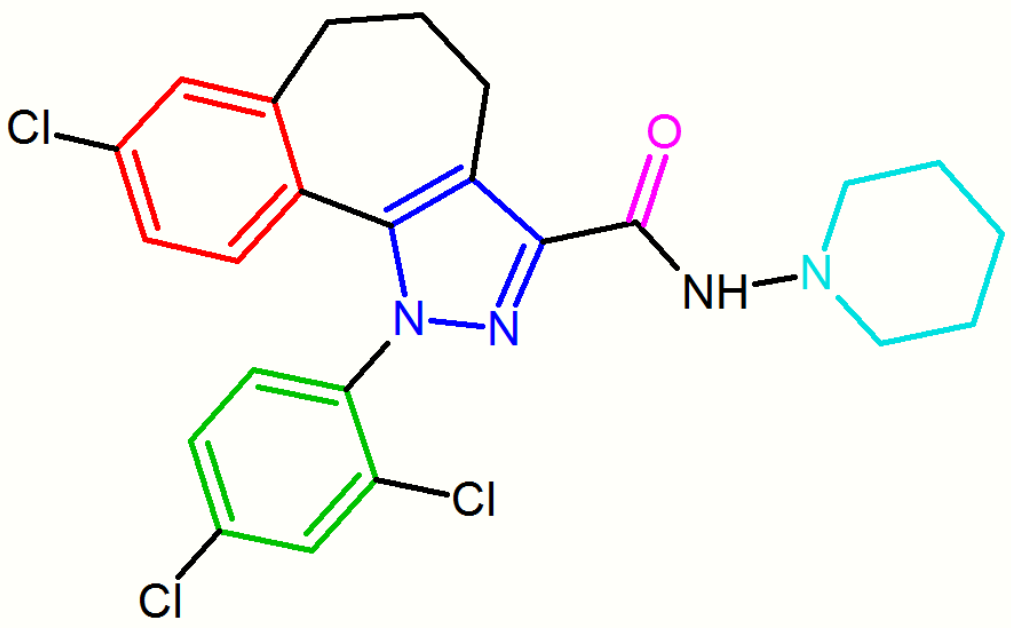
Several attempts have been made to increase the affinity of the diarylpyrazole derivatives by rigidifying the structure of rimonabant. In terms of the general pharmacophore model the units A, B and/or C are connected by additional bonds leading to rigid molecules. For example, the condensed polycyclic pyrazole NESS-0327 showed 5000 times more affinity for the CB1 receptor than rimonabant. However, this compound possesses a poor central bioavailability.[16][18]
Another compound, the indazole derivative O-1248, can be regarded as an analog of rimonabant wherein its 5-aryl group is fused to the pyrazole moiety. However, this structural modification resulted in a 67-fold decrease in CB1 receptor affinity.[18]
These diarylpyrazole derivatives of rimonabant are summarized in Table 1.
 |
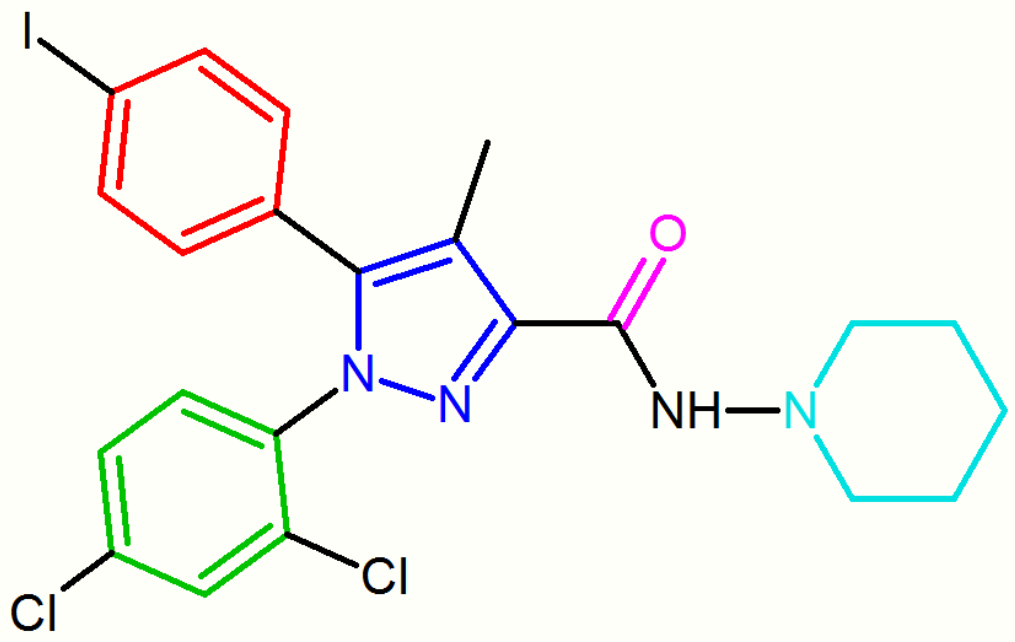 |
| SR147778 | AM251 |
 |
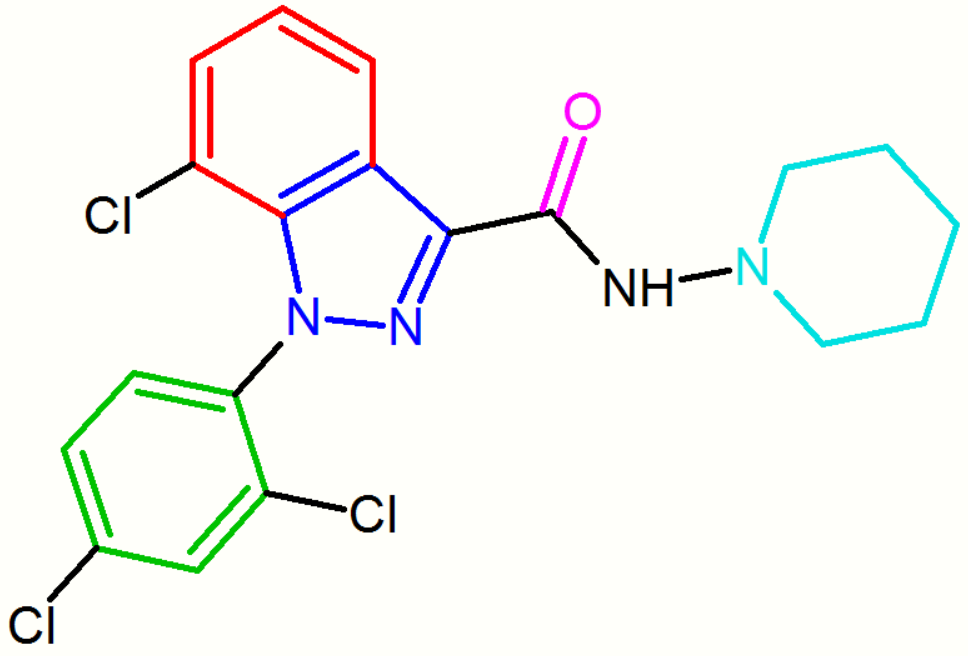 |
| NESS-0327 | O-1248 |
4.6. Other Derivatives
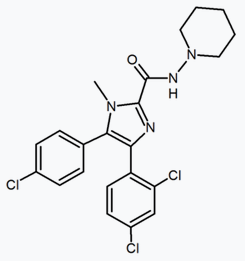
Structurally different from the 1,5-diarylpyrazoles are the chemical series of the 3,4-diarylpyrazolines. Within this series is SLV-319 (ibipinabant), a potent CB1 antagonist which is about 1000-fold more selective for CB1 compared with CB2 and displays in vivo activity similar to rimonabant.[18][24]
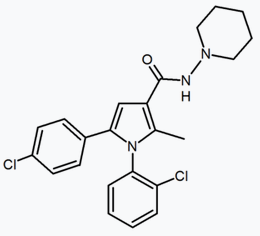
Another approach used to develop analogs of rimonabant was to replace central pyrazole ring by another heterocycle. An example of this approach are 4,5-diarylimidazoles and 1,5-diarylpyrrole-3-carboxamides.[24]
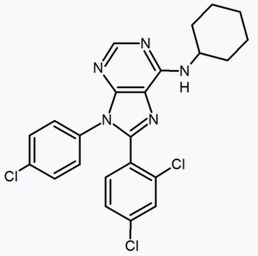
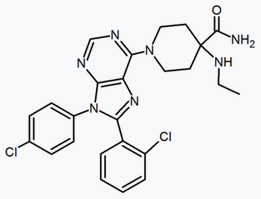
A large number of fused bicyclic derivatives of diaryl-pyrazole and imidazoles have been reported. An example of these is a purine derivative where a pyrimidine ring is fused to an imidazole ring.[24] Otenabant (CP-945,598) is an example of a fused bicyclic derivative developed by Pfizer.[26]
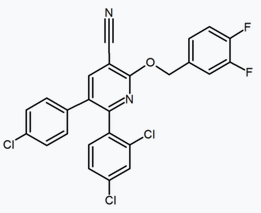
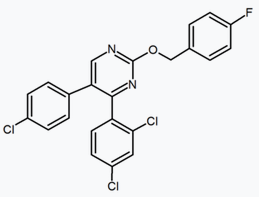
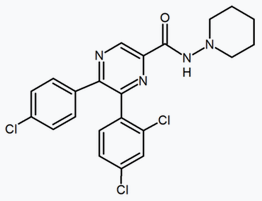
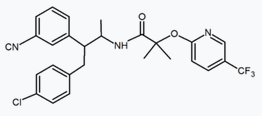
Several research groups have studied six-membered ring pyrazole bioisosteres. For example, one 2,3-diarylpyridine derivative was shown to be potent and selective CB1 inverse agonist. The structure of this compound demonstrates the possibility that the amide moiety of rimonabant could be split into a lipophilic (benzyloxy) and a polar (nitrile) functionality. Other six-membered ring analogs are for example pyrimidines and pyrazines.[24]
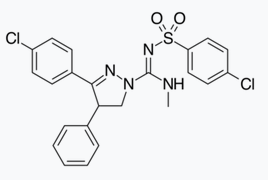
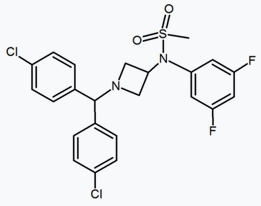
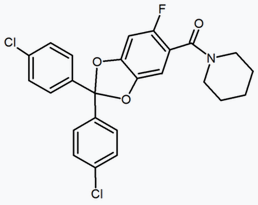
In addition to the five and six-membered ring analogs there are other cyclic derivatives such as the azetidines. One example is the methylsulfonamide azetidine derivative which has a 1,1-diaryl group that mimics the 1,5-diaryl moiety of the diarylpyrazoles. The sulfonyl group serves as a hydrogen bond acceptor. The 1,1-diaryl group is also present in derivatives such as the benzodioxoles and hydantoins.[18][24]
Acyclic analogs have also been reported. These analogs contain a 1,2-diaryl motif which corresponds to the 1,5-diaryl substituents of rimonabant.[24] An example of an acyclic analog is taranabant (MK-0364) developed by Merck.[26]
Representatives of these analogs are summarized in Table 2.
 |
 |
 |
|
| Type of derivative |
3,4-Diarylpyrazoline (Ibipinabant) | 4,5-Diarylimidazole | 1,5-Diarylpyrrole-3-carboxamides |
 |
 |
 |
|
| Type of derivative |
Purine (pyrimidine ring fused to an imidazole ring) |
Purine derivative (Otenabant) | 2,3-Diarylpyridine |
 |
 |
 |
|
| Type of derivative |
Pyrimidine | Pyrazine | Methylsulfonamide azetidine |
 |
 |
 |
|
| Type of derivative |
Benzodioxole | Hydantoin | Acyclic derivative (Taranabant) |
4.7. CB1 Receptor Antibodies
Antibodies against the CB1 receptor have been developed and introduced into clinical use in Russia .[27] They include brizantin (Russian: Бризантин®) and dietressa (Russian: Диетресса®).[27] Brizantin is indicated for the treatment of nicotine withdrawal and smoking cessation and dietressa is indicated for weight loss.[27] Dietressa is available over-the-counter in Russia.[27][2]
5. Current Status
Rimonabant (Acomplia) has been approved in the European Union (EU) since June 2006 for the treatment of obesity. On 23 October 2008 the European Medicines Agency (EMEA) has recommended the suspension of the marketing authorization across the EU for Acomplia from Sanofi-Aventis based on the risk of serious psychiatric disorders.[28] On 5 November 2008 Sanofi-Aventis announced discontinuation of rimonabant clinical development program.[29]
Sanofi-Aventis has also discontinued development of surinabant (SR147778), a CB1 receptor antagonist for smoking cessation (31 October 2008).[30]
Merck has stated in its press release on 2 October 2008 that they will not seek regulatory approval for taranabant (MK-0364) to treat obesity and will discontinue its Phase III clinical development program. Data from Phase III clinical trial showed that greater efficacy and more adverse effects were associated with the higher doses of taranabant and it was determined that the overall profile of taranabant does not support further development for obesity.[31]
Another pharmaceutical company, Pfizer, terminated the Phase III development program for its obesity compound otenabant (CP-945,598), a selective antagonist of the CB1 receptor. According to Pfizer their decision was based on changing regulatory perspectives on the risk/benefit profile of the CB1 class and likely new regulatory requirements for approval.[32]
A number of initiatives have been published to develop CB1 antagonists that target only peripheral CB1 receptors by restricting their ability to cross the blood brain barrier. Among these initiatives 7TM Pharma has reported the development of TM38837. A review has now published on the approaches and compounds being pursued as peripherally restricted CB1 receptor blockers.[33]
References
- Pertwee, R.G. (2006), "Cannabinoid pharmacology: the first 66 years", British Journal of Pharmacology 147 (S1): S163–S171, doi:10.1038/sj.bjp.0706406, PMID 16402100 http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1760722
- Reggio, P.H. (2003), "Pharmacophores for Ligand Recognition and Activation/Inactivation of the Cannabinoid Receptors", Current Pharmaceutical Design 9 (20): 1607–1633, doi:10.2174/1381612033454577, PMID 12871061, http://science.kennesaw.edu/projects/cab/CurPharmDes-Reggio-2003.pdf
- Schlesinger S. Untersuchung der Cannabis sativa. Repertorium für die Pharmacie. 1840:190-208.
- Decourtive E. Note sur le haschisch. CR Hebd Séances Acad Sci..1848;26:509-510.
- "Archived copy". http://www.dialogues-cns.com/brochures/35/pdf/35.pdf#page=61.
- Howlett, A.C.; Breivogel, C.S.; Childers, S.R.; Deadwyler, S.A.; Hampson, R.E.; Porrino, L.J. (2004), "Cannabinoid physiology and pharmacology: 30 years of progress", Neuropharmacology 47: 345–358, doi:10.1016/j.neuropharm.2004.07.030, PMID 15464149 https://dx.doi.org/10.1016%2Fj.neuropharm.2004.07.030
- Patel, P.N.; Pathak, R. (2007), "Rimonabant: A novel selective cannabinoid – 1 receptor antagonist for treatment of obesity", American Journal of Health-System Pharmacy 64 (5): 481–489, doi:10.2146/060258, PMID 17322160 https://dx.doi.org/10.2146%2F060258
- Barth, F.; Rinaldi-Carmona, M. (1999), "The Development of Cannabinoid Antagonists", Current Medicinal Chemistry 6 (8): 745–755, PMID 10469889, http://www.bentham.org/cmc/contabs/vol6-8.html
- Mackie K (2006), "Cannabinoid receptors as therapeutic targets", Annu. Rev. Pharmacol. Toxicol. 46: 101–22, doi:10.1146/annurev.pharmtox.46.120604.141254, PMID 16402900, http://www.safeaccessnow.org/downloads/Mackie%20CB%20Receptors%20Annur%20Rev%20Pharm%20Tox%202006.pdf.
- Fong TM, Heymsfield SB (September 2009), "Cannabinoid-1 receptor inverse agonists: current understanding of mechanism of action and unanswered questions", Int J Obes (Lond) 33 (9): 947–55, doi:10.1038/ijo.2009.132, PMID 19597516. https://dx.doi.org/10.1038%2Fijo.2009.132
- Svíženská, I.; Dubový, P.; Šulcová, A. (2008), "Cannabinoid receptor 1 and 2 ( CB1 and CB2), their distribution, ligands and functional involvement in nervous system structures – A short review", Pharmacology Biochemistry and Behavior 90 (4): 501–511, doi:10.1016/j.pbb.2008.05.010, PMID 18584858 https://dx.doi.org/10.1016%2Fj.pbb.2008.05.010
- Xie, S.; Furjanic, M.A.; Ferrara, J.J.; McAndrew, N.R.; Ardino, E.L.; Ngondara, A.; Bernstein, Y.; Thomas, K.J. et al. (2007), "The endocannabinoid system and rimonabant: a new drug with a novel mechanism of action involving cannabinoid CB1 receptor antagonism – or inverse agonism – as potential obesity treatment and other therapeutic use", Journal of Clinical Pharmacy and Therapeutics 32 (3): 209–231, doi:10.1111/j.1365-2710.2007.00817.x, PMID 17489873 https://dx.doi.org/10.1111%2Fj.1365-2710.2007.00817.x
- Ashton, J.S.; Wright, J.L.; McPartland, J.M.; Tyndall, J.D.A. (2008), "Cannabinoid CB1 and CB2 Receptor Ligand Specificity and the Development of CB2-Selective Agonists", Current Medicinal Chemistry 15 (14): 1428–1443, doi:10.2174/092986708784567716, PMID 18537620 https://dx.doi.org/10.2174%2F092986708784567716
- Di Marzo, V. (2008), "CB1 receptor antagonism: biological basis for metabolic effects", Drug Discovery Today 13 (23–24): 1–16, doi:10.1016/j.drudis.2008.09.001, PMID 18824122 https://dx.doi.org/10.1016%2Fj.drudis.2008.09.001
- Rinaldi – Carmona, M.; Barth, F.; Héaulme, M.; Shire, D.; Calandra, B.; Congy, C.; Martinez, S.; Maruani, J. et al. (1994), "SR141716A, a potent and selective antagonist of the brain cannabinoid receptor", FEBS Letters 350 (2–3): 240–244, doi:10.1016/0014-5793(94)00773-X, PMID 8070571 https://dx.doi.org/10.1016%2F0014-5793%2894%2900773-X
- Muccioli, G.G.; Lambert, D.M. (2005), "Current Knowledge on the Antagonists and Inverse Agonists of Cannabinoid Receptors", Current Medicinal Chemistry 12 (12): 1361–1394, doi:10.2174/0929867054020891, PMID 15974990, http://www.farm.ucl.ac.be/Full-texts-FARM/Muccioli-2005-2.pdf
- Ortega-Gutiérrez, S.; López-Rodriguez, M.L. (2005), "CB1 and CB2 Cannabinoid Receptor Binding Studies Based on Modeling and Mutagenesis Approaches", Mini-Reviews in Medicinal Chemistry 5 (7): 651–658, doi:10.2174/1389557054368754, PMID 16026311 https://dx.doi.org/10.2174%2F1389557054368754
- Lange, Jos H.M.; Kruse, Chris G. (2005), "Medicinal chemistry strategies to CB1 cannabinoid receptor antagonists", Drug Discovery Today 10 (10/24): 693–702, doi:10.1016/S1359-6446(05)03427-6, PMID 15896682 https://dx.doi.org/10.1016%2FS1359-6446%2805%2903427-6
- McAllister, S.D.; Rizvi, G.; Anavi-Goffer, S.; Hurst, D.P.; Barnett-Norris, J.; Lynch, D.L.; Reggio, P.H.; Abood, M.E. (2003), "An Aromatic Microdomain at the Cannabinoid CB1 Receptor Constitutes an Agonist/Inverse Agonist Binding Region", Journal of Medicinal Chemistry 46 (24): 5139–5152, doi:10.1021/jm0302647, PMID 14613317 https://dx.doi.org/10.1021%2Fjm0302647
- Fan, H.; Kotsikorou, E.; Hoffman, A.F.; Ravert, H.T.; Holt, D.; Hurst, D.P.; Lupica, C.R.; Reggio, P.H. et al. (2008), "Analogs of JHU75528, a PET ligand for imaging of cerebral cannabinoid receptors (CB1): Development of ligands with optimized lipophilicity and binding affinity", European Journal of Medicinal Chemistry 44 (2): 1–16, doi:10.1016/j.ejmech.2008.03.040, PMID 18511157 http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2728551
- Foloppe, N.; Allen, N.H.; Bentlev, C.H.; Brooks, T.D.; Kennett, G.; Knight, A.R.; Leonardi, S.; Misra, A. et al. (2008), "Discovery of novel class of selective human CB1 inverse agonists", Bioorganic & Medicinal Chemistry Letters 18 (3): 1199–1206, doi:10.1016/j.bmcl.2007.11.133, PMID 18083560 https://dx.doi.org/10.1016%2Fj.bmcl.2007.11.133
- Jagerovic, N.; Fernandez – Fernandez, C.; Goya, P. (2008), "CB1 Cannabinoid Antagonists: Structure – Activity Relationships and Potential Therapeutic Applications", Current Topics in Medicinal Chemistry 8 (3): 205–230, doi:10.2174/156802608783498050, PMID 18289089 https://dx.doi.org/10.2174%2F156802608783498050
- "AM-251 and rimonabant act as direct antagonists at mu-opioid receptors: implications for opioid/cannabinoid interaction studies.", Neuropharmacology. 2012 Oct;63(5):905-15. doi: 10.1016/j.neuropharm.2012.06.046. Epub 2012 Jul 4. https://www.sciencedirect.com/science/article/pii/S0028390812003048?via%3Dihub
- Barth, F. (2005), "CB1 Cannabinoid Receptor Antagonists", Annual Reports in Medicinal Chemistry Volume 40, Annual Reports in Medicinal Chemistry, 40, pp. 103–118, doi:10.1016/S0065-7743(05)40007-X, ISBN 978-0-12-040540-4, http://www.elsevier.com/wps/find/bookdescription.cws_home/706482/description#description, retrieved 2008-11-14
- Vemuri, V.K.; Janero, D.R.; Makriyannis, A. (2008), "Pharmacotherapeutic targeting of the endocannabinoid signaling system: Drugs for obesity and the metabolic syndrome", Physiology & Behavior 93 (4–5): 671–686, doi:10.1016/j.physbeh.2007.11.012, PMID 18155257 http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3681125
- Kim, M.; Yun, H.; Kwak, H.; Kim, J.; Lee, J. (2008), "Design, chemical synthesis, and biological evaluation of novel triazolyl analogues of taranabant (MK-0364), a cannabinoid-1 receptor inverse agonist", Tetrahedron 64 (48): 10802–10809, doi:10.1016/j.tet.2008.09.057 https://dx.doi.org/10.1016%2Fj.tet.2008.09.057
- Barchukov, V. V.; Zhavbert, E. S.; Dugina, Yu. L.; Epstein, O. I. (2015). "The Use of Release-Active Antibody-Based Preparations for Vertigo Prevention in Adults". Bulletin of Experimental Biology and Medicine 160 (1): 61–63. doi:10.1007/s10517-015-3098-z. ISSN 0007-4888. PMID 26608378. https://dx.doi.org/10.1007%2Fs10517-015-3098-z
- [1]
- "Sanofi, a global biopharmaceutical company focused on human health - Sanofi". http://en.sanofi-aventis.com/binaries/20081105_rimonabant_en_tcm28-22682.pdf.
- http://www.sanofi-aventis.ca/live/ca/medias/DA924A61-8E54-4D1D-B1FB-71C953E398C2.pdf
- "Archived copy". http://www.merck.com/newsroom/press_releases/research_and_development/2008_1002_print.html.
- http://www.pfizer.com/news/press_releases/pfizer_press_releases.jsp?rssUrl=http://mediaroom.pfizer.com/portal/site/pfizer/index.jsp?ndmViewId=news_view&ndmConfigId=1010794&newsId=20081105006339&newsLang=en
- Chorvat, Robert J. (2013). "Peripherally restricted CB-1 receptor blockers". Bioorg. Med. Chem. Lett. 23 (17): 4751–4760. doi:10.1016/j.bmcl.2013.06.066. PMID 23902803. https://dx.doi.org/10.1016%2Fj.bmcl.2013.06.066

