 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | TERESA GASULL | + 2573 word(s) | 2573 | 2020-12-03 03:48:05 | | | |
| 2 | Vivi Li | + 888 word(s) | 3461 | 2020-12-04 05:00:14 | | |
Video Upload Options
Death-associated protein kinase 1 (DAPK1) is a pleiotropic hub of a number of networked distributed intracellular processes. Among them, DAPK1 is known to interact with the excitotoxicity driver NMDA receptor (NMDAR), and in sudden pathophysiological conditions of the brain, e.g., stroke, several lines of evidence link DAPK1 with the transduction of glutamate-induced events that determine neuronal fate. In turn, DAPK1 expression and activity are known to be affected by the redox status of the cell. To delineate specific and differential neuronal DAPK1 interactors in stroke-like conditions in vitro, we exposed primary cultures of rat cortical neurons to oxygen/glucose deprivation (OGD), a condition that increases reactive oxygen species (ROS) and lipid peroxides. OGD or control samples were co-immunoprecipitated separately, trypsin-digested, and proteins in the interactome identified by high-resolution LC-MS/MS. Data were processed and curated using bioinformatics tools. OGD increased total DAPK1 protein levels, cleavage into shorter isoforms, and dephosphorylation to render the active DAPK1 form. The DAPK1 interactome comprises some 600 proteins, mostly involving binding, catalytic and structural molecular functions. OGD up-regulated 190 and down-regulated 192 candidate DAPK1-interacting proteins. Some differentially up-regulated interactors related to NMDAR and specially the interaction of DAPK1 with the protein leucine-rich repeat of flightless interacting protein 1 (LRRFIP1), that we observed upregulated during OGD and ferroptosis , were validated by WB.
1. Introduction
Modern biological research reveals that, in order to regulate essential cellular functions in a given condition, the cell sets its proteome into highly complex assemblies of multiproteins. In this regard, death-associated protein kinase 1 (DAPK1) is an emerging hub kinase involved in a number of cellular functions, both in physiological and pathophysiological conditions. While it has been associated to excitotoxic neuronal damage, its role in ischemia is still poorly studied.
A relationship between reactive oxygen species (ROS) and DAPK1 has been observed. Thus, in tumor cell lines, it has been described that antioxidants increase the expression of DAPK1 [1], and that ROS facilitate protein phosphatase 2A (PP2A)-mediated dephosphorylation of pDAPK1 to render the active, non-phosphorylated, DAPK1 form [2]. In the brain, it has been recently reported that ischemia downregulates miR-98-5p, and that experimentally upregulated miR-98-5p in stroked mice inhibits ROS production, reduces infarction and suppresses DAPK1 signaling [3].
During ischemia, the excess glutamate released by presynaptic glutamatergic terminals spills over the synaptic cleft to interact with and signal downstream of the extrasynaptic N-methyl-d-aspartate (NMDA) receptor (NMDAR) and also increases the presence of ROS-modified lipids in the cell membrane. NMDAR at the neuronal membrane is pivotal for normal cell activity and neurotransmission, but its overactivation plays a principal role in neurodegeneration through excitotoxicity. As direct antagonism of NMDAR has been revealed to be poorly tolerated in the clinical arena so far, other therapeutic strategies have been postulated to prevent the association of the NMDAR with intracellular protein signaling transducers networking for excitotoxicity. Specifically, the cytoplasmic C-terminal domain (CTD) of the NMDAR 2B subunit (NR2B), which in neurons is located mostly extrasynaptically, has been proposed as a good therapeutic target for this purpose, based on the fact that CTD directly senses Ca2+ entry through NMDAR. Replacing the CTD of NR2B with that of 2A subunit-containing NMDAR (NR2A) by targeted exon exchange has been shown to reduce vulnerability to excitotoxicity [4]. Additionally, NR2B-containing NMDAR have been identified, and recently retested and confirmed, to be primary mediators of ischemic stroke damage [5][6]. Therefore, although other intracellular mechanisms are involved in excitotoxicity, CTD-coupled events are thought to be the first downstream excitotoxicity intracellular signals towards cell death.
While the mechanisms of CTD-NR2B-mediated excitotoxicity still remain largely unexplained, it is worth highlighting that: (1) the CTD-postsynaptic density (PSD) 95-neuronal nitric oxide synthase (nNOS) or CTD-DAPK1 hubs are involved in excitotoxicity, as shown by protection exerted by genetic knockdowns [7][8]; (2) very recently, other authors identified antagonistic effects of ischemia-regulated endogenous miRNAs (AK038897 and miR-26a-5p) on DAPK1 that finally affect cerebral ischemia/reperfusion injury [9]; and (3) mice carrying a specific mutation that prevents binding of DAPK1 to NR2B are protected against stroke damage by inhibiting injurious Ca2+ influx through extrasynaptic NMDAR channels without altering synaptic NMDAR functions [5]. While the absence of DAPK1 has been shown to protect neurons from a variety of acute insults, the true requirement of DAPK1 for the excitotoxic signaling to proceed has also been questioned [10].
Some pioneering studies revealed that DAPK1 is the most prevalent protein recruited to the cytoplasmic tail of NR2B during cerebral ischemia [8], with virtually no interaction with NR2A. During cerebral ischemia, DAPK1 is dephosphorylated and then interacts and phosphorylates the CTD-NR2B to enhance the inflow of Ca2+ into the neuron, further exacerbating excitotoxicity in a vicious circle [11]. The fact that this effect of DAPK1 on the increase of Ca2+ entry has not been observed through synaptic, mostly NR2A, NMDAR [8][12] reinforces the idea that the reciprocal DAPK1 interaction with NR2B is specific and differential, and implies detrimental, if not dramatic, functional consequences for the neuron.
2. Discussion
DAPK1 has been reported to be a main actor in the highly complex intracellular hubs driving neurodegeneration and/or survival organized around the intracellular NR2B-CTD signaling through CTD-CaMKII [13][14], CTD-PSD95-nNOS [7], and CTD-DAPK1 itself [8]. The body of knowledge of the last one is still scarce, probably because it was the last to be introduced, and further complicated by, at least, the following facts: 1/OGD increases DAPK1 levels preceding induction of neuronal death (Figure 1A–C), 2/OGD induces DAPK1 activation by dephosphorylation (Figure 1F) [8][15], and 3/proteolytic processing of DAPK1 by cathepsin around aa sequence 836–947 [11,25] generates 110 and 120 kDa bands, and a reduction of the full length DAPK1 form (Figure 1C,D). A previous study reported an OGD-induced increase of the full-length DAPK band in WB in vitro using a neuroblastoma cell line and longer exposure times to OGD [16]. These striking different conditions might account for the discrepancy. The cathepsin cleavage splits apart the DAPK1 kinase domain, located at the N-term and required to bind NR2B, from the canonical death domain located at the C-term.
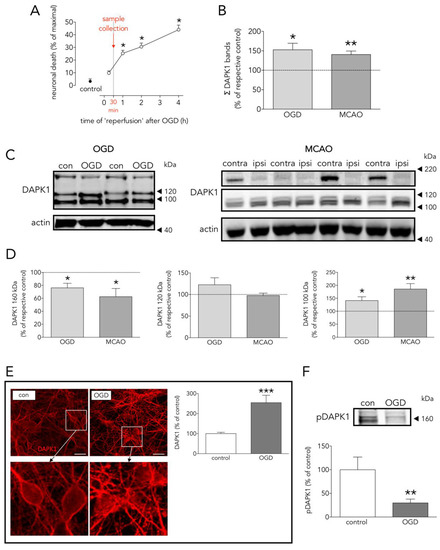
Figure 1. Effects of OGD on neuronal death, and DAPK1 expression, cleavage, and dephosphorylation. (A) Effect of time of ‘reperfusion’ after OGD on neuronal death; as depicted, experimental samples were taken at 30 min for further processing (one-way ANOVA plus SNK, n = at least 5 independent primary neuron culture preparations, with at least 3 technical replicates each). (B) Effect of OGD and MCAO on total DAPK1 levels (OGD: t test, n = 3 independent primary neuron culture preparations, with 3 technical replicates each; MCAO: paired t test, n = 9 rats, comparing ipsilateral (ipsi, ischemic) brain hemisphere with the contralateral (contra, control) one). (C) WB showing the effect of OGD or MCAO on DAPK1 bands. (D) Quantification of the effect of OGD or MCAO on DAPK1 cleavage and levels of the resulting bands (OGD: t test, n = 3 independent primary neuron culture preparations, with 3 technical replicates each; MCAO: paired t test, n = 9 rats, comparing ipsilateral (ipsi, ischemic) brain hemisphere with the contralateral (contra, control) one). (E) Effect of OGD on DAPK1 levels as measured by immunocytochemistry; insets in the upper pictures are shown magnified in the bottom pictures (t test, n = 3 independent primary neuron culture preparations, with 5 technical replicates each). (F) WB and quantification of the effect of OGD on pDAPK1 levels (t test, n = 3 independent primary neuron culture preparations, with 3 technical replicates each). Results are shown as the mean and SEM. * p < 0.05, ** p < 0.01, *** p < 0.005 vs. respective control in all graphs. Scale bar: 30 µm. Note that in (B), total DAPK1 levels represent protein expression.
To gain knowledge of new NR2B-CTD-related DAPK1 targets suitable to reduce excitotoxicity/ischemic-like damage, we used an affinity purification-mass spectrometry identification strategy. We used a specific and immunoprecipitation-qualified anti-DAPK1 antibody recognizing epitopes at the N-term, in the vicinity of the hotspot amino acid residues nearby the DAPK1 kinase domain [17]; as previously mentioned, this antibody demonstrated specific when tested in DAPK1 knock-out samples [8]. We found that our IP strategy resulted in full-length and truncated DAPK1 molecules. This was, in fact, observed by the OGD-induced increase in the relative levels of the DAPK1 100 kDa WB band, as compared with the full length DAPK1 molecule (160 kDa) pulled down in control samples (Figure 2B), as previously reported [11]. Truncated DAPK1 lacked a large fragment containing part of the cytoskeleton-binding region plus a phosphatase-binding site and the death domain.
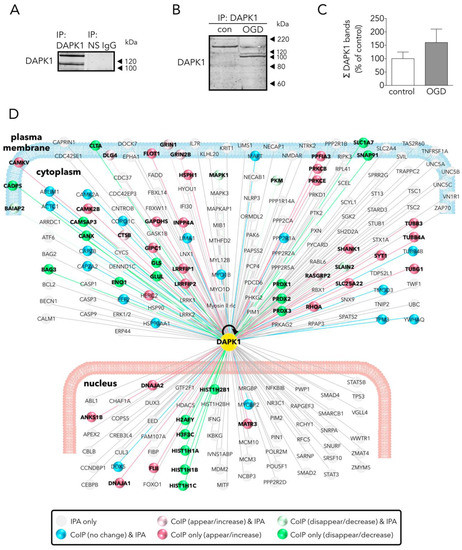
Figure 2. Main effects of OGD on the neuronal DAPK1 interactome. (A) WB shows that DAPK1 immunoprecipitates using a specific anti-DAPK1 antibody (IP: DAPK1, left lane), but it does not when using a non-specific antibody (IP: NS IgG, right lane). (B) WB showing the effect of OGD on the DAPK1 bands obtained by immunoprecipitation. (C) Graph showing the amount of DAPK1 obtained after immunoprecipitation of control and OGD samples (t test, n = 3 independent primary neuron culture preparations, with 3 technical replicates each). Note that, in this graph, differently than in WB in Figure 1B, DAPK1 data represent protein co-immunoprecipitated from neurons. (D) Drawing depicting some of the DAPK1 interactors found in the present study and those present in the IPA database; color codes of proteins are self-explained in the legend within the drawing. Results are shown as the mean and SEM.
We identified a total of 596 proteins interacting with the pulled down DAPK1 complex in control neurons (Table S1; some of these proteins are depicted in Figure 2D). A previous report, using IP with anti-NR2B antibodies on previously fractioned synaptic and extrasynaptic pools and then analyzed using proteomics, obtained similar figures (700 proteins) [18]. LC-MS differential proteomics allowed us to screen and identify proteins in the DAPK1 interactome that consistently appeared, disappeared, or showed >1.4-fold change in OGD-exposed as compared to control neurons (190 were found overrepresented and 192 underrepresented; see Tables S3 and S4, respectively). Figure 3 shows a detailed Panther™ classification of all DAPK1 protein interactors in control neurons and the increased interactors in OGD-exposed neurons. We observed that several cytoskeleton proteins change their degree of interaction in the DAPK1 interactome after OGD. In general terms, some microtubule- and actin-related molecules, and also phosphatases, are underrepresented or lacking, whereas other cytoskeleton proteins such as tubulins, myosins, tropomyosins and molecules related to intermediate filaments tend to be increased. These changes are consistent with the fact that DAPK1 cleavage truncates the molecule into two halves of similar length, each half retaining half of the cytoskeleton-binding region. In this regard, the DAPK1 N-term half pulled down by IP lacks the phosphatase-binding site and the death domain. It is also noteworthy mentioning that, as far as we are aware, ours is the first study reporting a protein-protein interaction between DAPK1 and several histones, and that these physical interactions weaken after OGD. This is interesting since it has been described that histones may play a role in orchestrating the neuronal transcriptional response to experimental stroke by MCAO, OGD [19][20][21], and other challenges to cell survival such as excitotoxicity or ferroptosis [22][23][24]. Notably, we have also found that peroxiredoxin (PRDX) 1, PRDX2 and PRDX3, members of a family of antioxidant enzymes of crucial physiological importance, interact with DAPK1 and that this degree of association decreases after OGD.
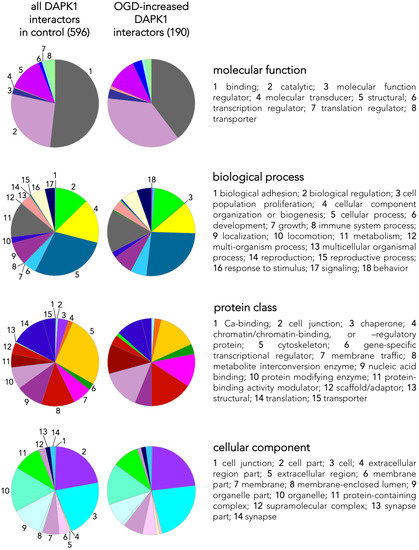
Figure 3. Classes and categorization of proteins in the DAPK1 interactome. Panther™ v15.0 (pantherdb.org) was used to classify proteins of the DAPK1 interactome in control neurons and those interactors found increased in OGD-exposed neurons (they are shown in a pie chart on the left). Four main classes are defined, each encompassing a number of different categories encoded by colors and explained on the right side of each class.
As expected by the reported increased association of DAPK1 to NR2B during ischemia [8], our IP plus LC results show that OGD increased NR2B in the neuronal DAPK1 interactome (Figure 2D) and confirmed by IP plus WB (Figure 4A,B). Additionally, levels of other known CTD-NR2B-interacting molecules increased after OGD, including 1/PSD95, a.k.a. discs, large homolog 4 (DLG4) [4], 2/dedicator of cytokinesis protein 3 (DOCK3) [25], 3/protein kinase C [26], 4/CaMKII (confirmed by IP plus WB, Figure 4A,B) [13], and 5/NR2B-specific binding molecule GIPC (confirmed by IP plus WB, Figure 4A,B), this being a PDZ scaffolding protein reported to preferentially stabilize NR2B-rich NMDA receptors [27]. We observed these changes in the DAPK1 interactome despite 30% of NR2B molecules undergo ischemia-induced truncation to give rise to a 115 kDa WB band (Figure 4C,D), in agreement with previous reports [28][29][30]. In fact, most of the NR2B pulled down in the multiprotein complex in OGD-exposed samples is the truncated 115 kDa NR2B form, previously reported to lack a portion of the C-term intracellular tail due to calpain proteolytic activity boosted by ischemia or glutamate [30][31][32].
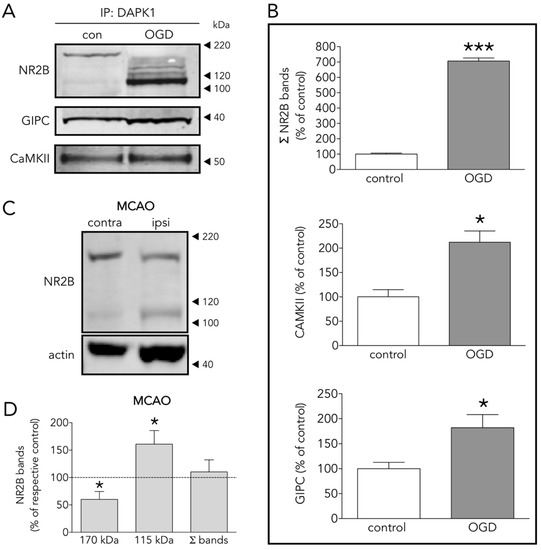
Figure 4. Several DAPK1 interactors are CTD-NR2B binding partners. (A) WB and (B) quantification of the effect of OGD on the co-immunoprecipitation of DAPK1 with the well-known DAPK1 interactors NR2B, CAMKII and GIPC (t test, n = 4–5 co-immunoprecipitations from independent primary neuron culture preparations). (C) WB showing the effect of MCAO on the NR2B band levels. (D) Effect of MCAO on 170 kDa, 115 kDa and total NR2B levels (paired t test, n = 9 rats, comparing ipsilateral (ipsi, ischemic) brain hemisphere with the contralateral (contra, control) one). The results are shown as the mean and SEM. * p < 0.05, *** p < 0.005 vs. respective control in all graphs.
The role of DAPK1 in ischemic excitotoxic damage has been so far associated to postsynaptic mechanisms of neurodegeneration. Nonetheless, very recent data suggest that DAPK1 might serve different roles at different locations of the neuron, since the DAPK1 interactor caytaxin, which we have also identified in the proteome of our DAPK1 Co-IP strategy (Table S1), has been reported to inhibit the catalytic activity of DAPK1 at presynaptic sites and to exert neuroprotective effects in stroke [33].
As one of the aims of the study was to find novel DAPK1 protein interactors differentially increased in the neuronal DAPK1 interactome after OGD, we focused on leucine-rich repeat of flightless I-interacting protein 1 (LRRFIP1), a.k.a. GC-binding factor 2 (GCF2), for further study due to several reasons. First, other proteins, previously identified as binding partners of LRRFIP1 in other reports, e.g., the molecules LRRFIP2 and flightless I homologue (FLII) [34][35], are also enriched in the OGD-DAPK1 interactome in our study. Second, and most important, LRRFIP1 has been previously observed upregulated in ischemia models, either in vivo or in cultured astrocytes [36][37]. In the rat, five transcripts have been identified so far that show upregulation in ischemia models [36][37]. These transcripts give rise to protein isoforms with predicted molecular masses of 83, 71, 48.9, 46.1 and 44.9, the last three isoforms displayed as a unique WB band of around 50 kDa. Furthermore, it has been reported that a prevalent polymorphism in the promoter of the glutamate transporter EAAT2 gene creates a new consensus binding site for LRRFIP1, which impairs glutamate uptake in astrocytes and increases the frequency of early neurological worsening in stroke [37]. Our findings in the DAPK1 interactome were further confirmed by the LRRFIP1/DAPK1 Co-IP ratio, which showed an increase of more than 50% (Figure 5A). Moreover, WB showed that OGD increased the expression of the major 83 and 50 kDa LRRFIP1 bands in neurons (Figure 5B,C). As far as we are aware, this is the first study reporting a direct LRRFIP1 interaction with DAPK1. In fact, several cross-IP assays (IP with antibodies against DAPK1 or LRRFIP1) revealed an association and coexistence of the 2 proteins after ischemia in adult rat brain or after OGD or NMDA treatment in primary culture neurons (Figure 6). Our results further suggest that LRRFIP1 might be playing an important role in ischemic brain damage and in OGD-treated neurons exposed to high oxidative stress conditions. In this regard, the potent natural antioxidant/inhibitor of oxidative stress resveratrol has been reported to increase LRRFIP1 levels in human peripheral blood mononuclear cells [38][39].
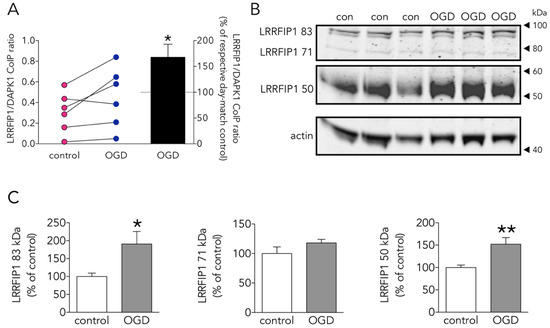
Figure 5. Effect of OGD on the new DAPK1-interacting candidate protein LRRFIP1. (A) Effect of OGD on the LRRFIP1/DAPK1 co-immunoprecipitation ratio (t test, n = 6 co-immunoprecipitations from independent primary neuron culture preparations). (B) WB showing the effect of OGD on the expression of different LRRFIP1 transcripts, and (C) graphs showing the quantification of this effect (83 and 50 kDa graphs: t test, and 71 kDa graph: Mann-Whitney U test, n = 3 independent primary neuron culture preparations, with 3 technical replicates each). Results in A (left panel) are shown as individual co-immunoprecipitation values, and in A (right panel) and C as the mean and SEM. * p < 0.05, ** p < 0.01 vs. respective control in all graphs.
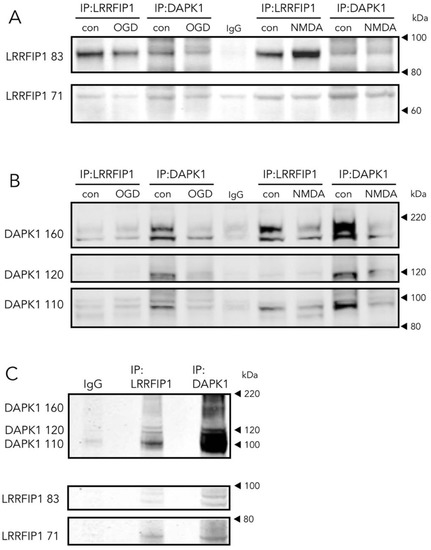
Figure 6. Direct and reverse co-immunoprecipitation of LRRFIP1 during ischemia in vivo and excitotoxicity in vitro. (A,B) WB showing the effect of OGD or NMDA on direct and reverse co-immunoprecipitation of LRRFIP1 and DAPK1 from rat cultured neurons. (C) WB showing direct and reverse co-immunoprecipitation of DAPK1 and LRRFIP1 from ischemic rat brain homogenates.
Beyond the increase in the expression of LRRFIP during OGD, inspection by confocal microscopy revealed that LRRFIP1 and NR2B colocalize at neurites, and also cell bodies, in control neurons, whereas both molecules are mostly evident at the soma after a brief NMDAR overactivation period (30 min) (Figure 7A) preceding excitotoxicity, a similar relocation effect that has also been observed for NR2B in previous reports [30][40][41]. Associated with this relocation of LRRFIP1, we also observed colocalization of LRRFIP1 with DAPK1 in the cell body of neurons undergoing nuclear pyknosis or fragmentation, but not in neurons shortly exposed to NMDA and showing normal morphology or in control neurons (Figure 7B). Our results might indicate a rearrangement within the neuron of LRRFIP1 molecules associated to NR2B or DAPK1 preceding excitotoxic neuronal death.
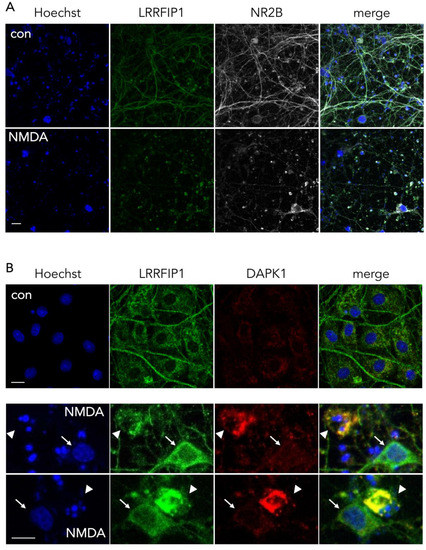
Figure 7. Representative ICC images showing the colocalization pattern of NR2B and DAPK1 with LRRFIP1. (A) Images showing the main cellular location of LRRFIP1 and NR2B in control and NMDA-treated neurons in vitro; scale bar: 30 µm. (B) Images showing the colocalization pattern of LRRFIP1 and DAPK1 in control neurons (upper panel, con), and in neurons with normal (arrows) or pyknotic (arrowheads) nucleus shortly after NMDA treatment (lower panels, NMDA); scale bars: 10 µm.
To further extend these results in order to know whether LRRFIP1 might be involved in other ROS-promoting neuronal slower death-inducing paradigms such as ferroptosis, we tested the ferroptosis inducer erastin in neuronal primary cultures. We observed that erastin (at concentrations that increase membrane lipid peroxides, not shown) increased levels of LRRFIP1 83 and 71 kDa bands, as measured by WB (Figure 8B,C), preceding neuronal death (Figure 8A); ICC showed increased total immunoreactive LRRFIP1 at 24 h at a higher erastin concentration (Figure 8D). Erastin acts mainly by inhibiting the cystine/glutamate antiporter system Xc- (composed of SLC7A11 and SLC3A2) [42][43], leading to cysteine starvation, glutathione depletion and deleterious excesses of cellular ROS. We observed that a 24-h exposure to the high 20 µM erastin concentration was enough to induce a significant increase of ferroptotic neuronal death, whereas longer exposure (72 h) was required to produce significant cell death at a lower erastin concentration (10 µM) (Figure 8A). We observed this increase in LRRFIP1 in mature neuron cultures at 24 h after 10–20 µM erastin with no change in levels of GPX4. A recent study reports erastin-induced reduction of cell viability associated to reduced levels of GPX4 [44], but the authors use very different conditions: immature 5 DIV neurons, exposed to higher erastin concentration (50 µM) and for 48 h. As impaired Xc- activity results in depletion of intracellular glutathione, an essential cofactor of GPX4, erastin would be expected to impair the enzymatic activity of GPX4, with a concomitant increase in ROS accumulation, even with no changes in protein GPX4 levels, as we have found (Figure 8B,C).
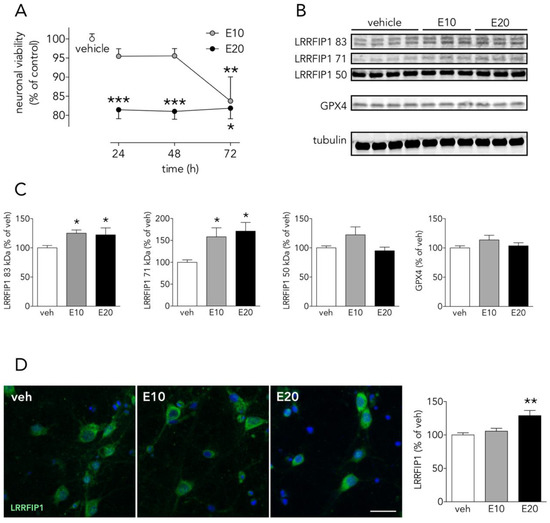
Figure 8. The ferroptosis inducer erastin increases neuronal LRRFIP1 levels. (A) Time-course effect of erastin on neuronal viability in vitro (one-way ANOVAs plus SNK, n = 3 independent primary neuron culture preparations, with 3 technical replicates each). (B) WB showing the effect of erastin on LRRFIP1 and GXP4 levels at 24 h, and (C) graphs depicting the quantification of this effect (one-way ANOVA plus SNK, n = 3 independent primary neuron culture preparations, with 3 technical replicates each). (D) Representative ICC images showing LRRFIP1 expression in neurons exposed to vehicle (veh) or erastin for 24 h, and a graph showing the quantification of this effect. Ten µM erastin: filled grey circles/bars or E10; 20 µM erastin: filled black circles/bars or E20. Results are shown as the mean and SEM. * p < 0.05, ** p < 0.01, *** p < 0.001 vs. vehicle in all graphs. Scale bar: 20 µm.
3. Conclusions
We have identified proteins up and down-regulated in the neuronal DAPK1-interactome by OGD, some of them related to NMDAR and validated by WB. We have identified for the first time LRRFIP1 as a DAPK1 partner, which we found up-regulated by OGD in the neuronal DAPK1 interactome, this being in line with the previously reported involvement of LRFFIP1 in ischemia. LRRFIP1 levels increased in neurons exposed to pro-oxidant conditions such as purely ferroptotic-induced cell death by exposure to erastin or to OGD-induced cell death, in which an excitotoxic and a ferroptotic-induced cell death component can converge. Therefore, the present study identifies LRRFIP1 as a novel partner of DAPK1, suitable as a target for neuroprotection following ischemic challenges.
References
- Zhou, Q.; Song, W.; Xiao, W. Dioscin induces demethylation of DAPK-1 and RASSF-1α genes via the antioxidant capacity, resulting in apoptosis of bladder cancer T24 cells. EXCLI J. 2017, 16, 101–112.
- Tsai, Y.T.; Chuang, M.J.; Tang, S.H.; Wu, S.T.; Chen, Y.C.; Sun, G.H.; Hsiao, P.W.; Huang, S.M.; Lee, H.J.; Yu, C.P.; et al. Novel cancer therapeutics with allosteric modulation of the mitochondrial C-Raf-DAPK complex by Raf inhibitor combination therapy. Cancer Res. 2015, 75, 3568–3582.
- Yu, S.; Zhai, J.; Yu, J.; Yang, Q.; Yang, J. miR-98-5p protects against cerebral ischemia/reperfusion injury through anti-apoptosis and anti-oxidative stress in mice. J. Biochem. 2020.
- Martel, M.A.; Ryan, T.J.; Bell, K.F.S.; Fowler, J.H.; McMahon, A.; Al-Mubarak, B.; Komiyama, N.H.; Horsburgh, K.; Kind, P.C.; Grant, S.G.N.; et al. The subtype of GluN2 C-terminal domain determines the response to excitotoxic insults. Neuron 2012, 74, 543–556.
- Tang, N.; Wu, J.; Zhu, H.; Yan, H.; Guo, Y.; Cai, Y.; Yan, H.; Shi, Y.; Shu, S.; Pei, L.; et al. Genetic mutation of GluN2B protects brain cells against stroke damages. Mol. Neurobiol. 2018, 55, 2979–2990.
- Hume, A.W.; Tasker, R.A. Endothelin-1-induced ischemic damage and functional impairment is mediated primarily by NR2B-containing NMDA receptors. Neurotox. Res. 2020, 37, 349–355.
- Aarts, M.; Liu, Y.; Liu, L.; Besshoh, S.; Arundine, M.; Gurd, J.W.; Wang, Y.T.; Salter, M.W.; Tymianski, M. Treatment of ischemic brain damage by perturbing NMDA receptor-PSD-95 protein interactions. Science 2002, 298, 846–850.
- Tu, W.; Xu, X.; Peng, L.; Zhong, X.; Zhang, W.; Soundarapandian, M.M.; Balel, C.; Wang, M.; Jia, N.; Zhang, W.; et al. DAPK1 interaction with NMDA receptor NR2B subunits mediates brain damage in stroke. Cell 2010, 140, 222–234.
- Wei, R.; Zhang, L.; Hu, W.; Wu, J.; Zhang, W. Long non-coding RNA AK038897 aggravates cerebral ischemia/reperfusion injury via acting as a ceRNA for miR-26a-5p to target DAPK1. Exp. Neurol. 2019, 314, 100–110.
- McQueen, J.; Ryan, T.J.; McKay, S.; Marwick, K.; Baxter, P.; Carpanini, S.M.; Wishart, T.M.; Gillingwater, T.H.; Manson, J.C.; Wyllie, D.J.A.; et al. Pro-death NMDA receptor signaling is promoted by the GluN2B C-terminus independently of Dapk1. Elife 2017, 6, 1–13.
- Shamloo, M.; Soriano, L.; Wieloch, T.; Nikolich, K.; Urfer, R.; Oksenberg, D. Death-associated protein kinase is activated by dephosphorylation in response to cerebral ischemia. J. Biol. Chem. 2005, 280, 42290–42299.
- Tovar, K.R.; Westbrook, G.L. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J. Neurosci. 1999, 19, 4180–4188.
- Bayer, K.U.; Schulman, H. CaM kinase: Still inspiring at 40. Neuron 2019, 103, 380–394.
- Omkumar, R.V.; Kiely, M.J.; Rosenstein, A.J.; Min, K.T.; Kennedy, M.B. Identification of a phosphorylation site for calcium/calmodulin-dependent protein kinase II in the NR2B subunit of the N-methyl-D-aspartate receptor. J. Biol. Chem. 1996, 271, 31670–31678.
- DeGregorio-Rocasolano, N.; Martí-Sistac, O.; Ponce, J.; Castelló-Ruiz, M.; Millán, M.; Guirao, V.; García-Yébenes, I.; Salom, J.B.; Ramos-Cabrer, P.; Alborch, E.; et al. Iron-loaded transferrin (Tf) is detrimental whereas iron-free Tf confers protection against brain ischemia by modifying blood Tf saturation and subsequent neuronal damage. Redox Biol. 2018, 15, 143–158.
- He, C.; Stroink, A.R.; Wang, C.X. The role of DAPK-BimEL pathway in neuronal death induced by oxygen-glucose deprivation. Neuroscience 2014, 258, 254–262.
- Tu, G.; Fu, T.; Yang, F.; Yao, L.; Xue, W.; Zhu, F. Prediction of GluN2B-CT1290-1310/DAPK1 interaction by protein-peptide docking and molecular dynamics simulation. Molecules 2018, 23, 3018.
- Swartzwelder, H.S.; Risher, M.L.; Miller, K.M.; Colbran, R.J.; Winder, D.G.; Wills, T.A. Changes in the adult GluN2B associated proteome following adolescent intermittent ethanol exposure. PLoS ONE 2016, 11, 1–18.
- Matheson, R.; Chida, K.; Lu, H.; Clendaniel, V.; Fisher, M.; Thomas, A.; Lo, E.H.; Selim, M.; Shehadah, A. Neuroprotective effects of selective inhibition of histone deacetylase 3 in experimental stroke. Transl. Stroke Res. 2020.
- Yildirim, F.; Ji, S.; Kronenberg, G.; Barco, A.; Olivares, R.; Benito, E.; Dirnagl, U.; Gertz, K.; Endres, M.; Harms, C.; et al. Histone acetylation and CREB binding protein are required for neuronal resistance against ischemic injury. PLoS ONE 2014, 9, e95465.
- Lanzillotta, A.; Pignataro, G.; Branca, C.; Cuomo, O.; Sarnico, I.; Benarese, M.; Annunziato, L.; Spano, P.F.; Pizzi, M. Targeted acetylation of NF-kappaB/RelA and histones by epigenetic drugs reduces post-ischemic brain injury in mice with an extended therapeutic window. Neurobiol. Dis. 2013, 49, 177–189.
- Zille, M.; Kumar, A.; Kundu, N.; Bourassa, M.W.; Wong, V.S.C.; Willis, D.; Karuppagounder, S.S.; Ratan, R.R. Ferroptosis in neurons and cancer cells is similar but differentially regulated by histone deacetylase inhibitors. Eneuro 2019, 6.
- Schlüter, A.; Aksan, B.; Fioravanti, R.; Valente, S.; Mai, A.; Mauceri, D. Histone deacetylases contribute to excitotoxicity-triggered degeneration of retinal ganglion cells in vivo. Mol. Neurobiol. 2019, 56, 8018–8034.
- Marin, C.; Langdon, C.; Alobid, I.; Fuentes, M.; Bonastre, M.; Mullol, J. Recovery of olfactory function after excitotoxic lesion of the olfactory bulbs is associated with increases in bulbar SIRT1 and SIRT4 expressions. Mol. Neurobiol. 2019, 56, 5643–5653.
- Namekata, K.; Kimura, A.; Kawamura, K.; Guo, X.; Harada, C.; Tanaka, K.; Harada, T. Dock3 attenuates neural cell death due to NMDA neurotoxicity and oxidative stress in a mouse model of normal tension glaucoma. Cell Death Differ. 2013, 20, 1250–1256.
- Liao, G.Y.; Wagner, D.A.; Hsu, M.H.; Leonard, J.P. Evidence for direct protein kinase-C mediated modulation of N-methyl-D-aspartate receptor current. Mol. Pharmacol. 2001, 59, 960–964.
- Yi, Z.; Petralia, R.S.; Fu, Z.; Swanwick, C.C.; Wang, Y.-X.; Prybylowski, K.; Sans, N.; Vicini, S.; Wenthold, R.J. The role of the PDZ protein GIPC in regulating NMDA receptor trafficking. J. Neurosci. 2007, 27, 11663–11675.
- Simpkins, K.L.; Guttmann, R.P.; Dong, Y.; Chen, Z.; Sokol, S.; Neumar, R.W.; Lynch, D.R. Selective activation induced cleavage of the NR2B subunit by calpain. J. Neurosci. 2003, 23, 11322–11331.
- Gascón, S.; Sobrado, M.; Roda, J.M.; Rodríguez-Peña, A.; Díaz-Guerra, M. Excitotoxicity and focal cerebral ischemia induce truncation of the NR2A and NR2B subunits of the NMDA receptor and cleavage of the scaffolding protein PSD-95. Mol. Psychiatry 2008, 13, 99–114.
- Wu, H.Y.; Yuen, E.Y.; Lu, Y.F.; Matsushita, M.; Matsui, H.; Yan, Z.; Tomizawa, K. Regulation of N-methyl-D-aspartate receptors by calpain in cortical neurons. J. Biol. Chem. 2005, 280, 21588–21593.
- Baudry, M.; Bi, X. Calpain-1 and calpain-2: The yin and yang of synaptic plasticity and neurodegeneration. Trends Neurosci. 2016, 39, 235–245.
- Cheng, S.Y.; Wang, S.C.; Lei, M.; Wang, Z.; Xiong, K. Regulatory role of calpain in neuronal death. Neural Regen. Res. 2018, 13, 556–562.
- Wang, S.; Chen, K.; Yu, J.; Wang, X.; Li, Q.; Lv, F.; Shen, H.; Pei, L. Presynaptic Caytaxin prevents apoptosis via deactivating DAPK1 in the acute phase of cerebral ischemic stroke. Exp. Neurol. 2020, 329, 113303.
- Goodall, A.H.; Burns, P.; Salles, I.; Macaulay, I.C.; Jones, C.I.; Ardissino, D.; De Bono, B.; Bray, S.L.; Deckmyn, H.; Dudbridge, F.; et al. Transcription profiling in human platelets reveals LRRFIP1 as a novel protein regulating platelet function. Blood 2010, 116, 4646–4656.
- Fong, K.S.K.; de Couet, H.G. Novel proteins interacting with the leucine-rich repeat domain of human flightless-I identified by the yeast two-hybrid system. Genomics 1999, 58, 146–157.
- Gubern, C.; Camós, S.; Hurtado, O.; Rodríguez, R.; Romera, V.G.; Sobrado, M.; Cañadas, R.; Moro, M.A.; Lizasoain, I.; Serena, J.; et al. Characterization of Gcf2/Lrrfip1 in experimental cerebral ischemia and its role as a modulator of Akt, mTOR and β-catenin signaling pathways. Neuroscience 2014, 268, 48–65.
- Mallolas, J.; Hurtado, O.; Castellanos, M.; Blanco, M.; Sobrino, T.; Serena, J.; Vivancos, J.; Castillo, J.; Lizasoain, I.; Moro, M.A.; et al. A polymorphism in the EAAT2 promoter is associated with higher glutamate concentrations and higher frequency of progressing stroke. J. Exp. Med. 2006, 203, 711–717.
- Giordo, R.; Nasrallah, G.K.; Al-Jamal, O.; Paliogiannis, P.; Pintus, G. Resveratrol inhibits oxidative stress and prevents mitochondrial damage induced by zinc oxide nanoparticles in zebrafish (Danio rerio). Int. J. Mol. Sci. 2020, 21, 3838.
- Tomé-Carneiro, J.; Larrosa, M.; Yáñez-Gascón, M.J.; Dávalos, A.; Gil-Zamorano, J.; Gonzálvez, M.; García-Almagro, F.J.; Ruiz Ros, J.A.; Tomás-Barberán, F.A.; Espín, J.C.; et al. One-year supplementation with a grape extract containing resveratrol modulates inflammatory-related microRNAs and cytokines expression in peripheral blood mononuclear cells of type 2 diabetes and hypertensive patients with coronary artery disease. Pharmacol. Res. 2013, 72, 69–82.
- Monnerie, H.; Hsu, F.C.; Coulter, D.A.; Le Roux, P.D. Role of the NR2A/2B subunits of the N-methyl-d-aspartate receptor in glutamate-induced glutamic acid decarboxylase alteration in cortical GABAergic neurons in vitro. Neuroscience 2010, 171, 1075–1090.
- Averna, M.; Pellegrini, M.; Cervetto, C.; Pedrazzi, M.; Bavestrello, M.; De Tullio, R.; Salamino, F.; Pontremoli, S.; Melloni, E. Physiological roles of calpain 1 associated to multiprotein NMDA receptor complex. PLoS ONE 2015, 10, 1–20.
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072.
- Dixon, S.J.; Patel, D.; Welsch, M.; Skouta, R.; Lee, E.; Hayano, M.; Thomas, A.G.; Gleason, C.; Tatonetti, N.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 2014, 2014, 1–25.
- Zhang, Y.; Fan, B.Y.; Pang, Y.L.; Shen, W.Y.; Wang, X.; Zhao, C.X.; Li, W.X.; Liu, C.; Kong, X.H.; Ning, G.Z.; et al. Neuroprotective effect of deferoxamine on erastin-induced ferroptosis in primary cortical neurons. Neural Regen. Res. 2020, 15, 1539–1545.


