 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Vivi Li | -- | 1375 | 2022-11-07 01:39:58 |
Video Upload Options
High-throughput sequencing technologies have led to a dramatic decline of genome sequencing costs and to an astonishingly rapid accumulation of genomic data. These technologies are enabling ambitious genome sequencing endeavours, such as the 1000 Genomes Project and 1001 (Arabidopsis thaliana) Genomes Project. The storage and transfer of the tremendous amount of genomic data have become a mainstream problem, motivating the development of high-performance compression tools designed specifically for genomic data. A recent surge of interest in the development of novel algorithms and tools for storing and managing genomic re-sequencing data emphasizes the growing demand for efficient methods for genomic data compression.
1. General Concepts
While standard data compression tools (e.g., zip and rar) are being used to compress sequence data (e.g., GenBank flat files), this approach has been criticized to be extravagant because genomic sequences often contain repetitive content (e.g., microsatellite sequences) or many sequences exhibit high levels of similarity (e.g., multiple genome sequences from the same species). Additionally, the statistical and information-theoretic properties of genomic sequences can potentially be exploited for compressing sequencing data.[1][2][3]
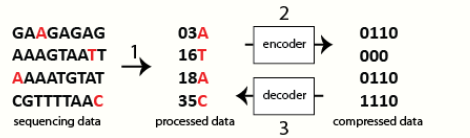
1.1. Base Variants
With the availability of a reference template, only differences (e.g., single nucleotide substitutions and insertions/deletions) need to be recorded, thereby greatly reducing the amount of information to be stored. The notion of relative compression is obvious especially in genome re-sequencing projects where the aim is to discover variations in individual genomes. The use of a reference single nucleotide polymorphism (SNP) map, such as dbSNP, can be used to further improve the number of variants for storage.[4]
1.2. Relative Genomic Coordinates
Another useful idea is to store relative genomic coordinates in lieu of absolute coordinates.[4] For example, representing sequence variant bases in the format ‘Position1Base1Position2Base2…’, ‘123C125T130G’ can be shortened to ‘0C2T5G’, where the integers represent intervals between the variants. The cost is the modest arithmetic calculation required to recover the absolute coordinates plus the storage of the correction factor (‘123’ in this example).
1.3. Prior Information about the Genomes
Further reduction can be achieved if all possible positions of substitutions in a pool of genome sequences are known in advance.[4] For instance, if all locations of SNPs in a human population are known, then there is no need to record variant coordinate information (e.g., ‘123C125T130G’ can be abridged to ‘CTG’). This approach, however, is rarely appropriate because such information is usually incomplete or unavailable.
1.4. Encoding Genomic Coordinates
Encoding schemes are used to convert coordinate integers into binary form to provide additional compression gains. Encoding designs, such as the Golomb code and the Huffman code, have been incorporated into genomic data compression tools.[5][6][7][8][9][10] Of course, encoding schemes entail accompanying decoding algorithms. Choice of the decoding scheme potentially affects the efficiency of sequence information retrieval.
2. Algorithm Design Choices
A universal approach to compressing genomic data may not necessarily be optimal, as a particular method may be more suitable for specific purposes and aims. Thus, several design choices that potentially impacts compression performance may be important for consideration.
2.1. Reference Sequence
Selection of a reference sequence for relative compression can affect compression performance. Choosing a consensus reference sequence over a more specific reference sequence (e.g., the revised Cambridge Reference Sequence) can result in higher compression ratio because the consensus reference may contain less bias in its data.[4] Knowledge about the source of the sequence being compressed, however, may be exploited to achieve greater compression gains. The idea of using multiple reference sequences has been proposed.[4] Brandon et al. (2009)[4] alluded to the potential use of ethnic group-specific reference sequence templates, using the compression of mitochondrial DNA variant data as an example (see Figure 2). The authors found biased haplotype distribution in the mitochondrial DNA sequences of Africans, Asians, and Eurasians relative to the revised Cambridge Reference Sequence. Their result suggests that the revised Cambridge Reference Sequence may not always be optimal because a greater number of variants need to be stored when it is used against data from ethnically distant individuals. Additionally, a reference sequence can be designed based on statistical properties [1][4] or engineered [11][12] to improve the compression ratio.
2.2. Encoding Schemes
The application of different types of encoding schemes have been explored to encode variant bases and genomic coordinates.[4] Fixed codes, such as the Golomb code and the Rice code, are suitable when the variant or coordinate (represented as integer) distribution is well defined. Variable codes, such as the Huffman code, provide a more general entropy encoding scheme when the underlying variant and/or coordinate distribution is not well-defined (this is typically the case in genomic sequence data).
3. List of Genomic Re-sequencing Data Compression Tools
The compression ratio of currently available genomic data compression tools ranges between 65-fold and 1,200-fold for human genomes.[4][5][6][7][8][9][10][13] Very close variants or revisions of the same genome can be compressed very efficiently (for example, 18,133 compression ratio was reported [6] for two revisions of the same A. thaliana genome, which are 99.999% identical). However, such compression is not indicative of the typical compression ratio for different genomes (individuals) of the same organism. The most common encoding scheme amongst these tools is Huffman coding, which is used for lossless data compression.
| Software | Description | Compression Ratio | Data Used for Evaluation | Approach/Encoding Scheme | Link | Use Licence | Reference |
|---|---|---|---|---|---|---|---|
| Genozip | A universal compressor for genomic files - compresses FASTQ, SAM/BAM/CRAM, VCF/BCF, FASTA, GVF, Phylip and 23andMe files | 60% to 99% | Human genome sequences from the 1000 Genomes Project | Genozip extensible framework | http://genozip.com | Free for non-commercial use | [14] |
| Genomic Squeeze (G-SQZ) | Lossless compression tool designed for storing and analyzing sequencing read data | 65% to 76% | Human genome sequences from the 1000 Genomes Project | Huffman coding | http://public.tgen.org/sqz | -Undeclared- | [8] |
| CRAM (part of SAMtools) | Highly efficient and tunable reference-based compression of sequence data | [15] | European Nucleotide Archive | deflate and rANS | http://www.ebi.ac.uk/ena/software/cram-toolkit | Apache-2.0 | [16] |
| Genome Compressor (GeCo) | A tool using a mixture of multiple Markov models for compressing reference and reference-free sequences | Human nuclear genome sequence | Arithmetic coding | http://bioinformatics.ua.pt/software/geco/ or https://pratas.github.io/geco/ | GPLv3 | [13] | |
| PetaSuite | Lossless compression tool for BAM and FASTQ files | 60% to 90% | Human genome sequences from the 1000 Genomes Project | https://www.petagene.com | Commercial | [17] | |
| GenomSys codecs | Lossless compression of BAM and FASTQ files into the standard format ISO/IEC 23092[18] (MPEG-G) | 60% to 90% | Human genome sequences from the 1000 Genomes Project | Context-adaptive binary arithmetic coding (CABAC) | https://www.genomsys.com | Commercial | [19] |
| Genie | Transcoding between the FASTA, FASTQ and SAM/BAM formats and the ISO/IEC 23092 [20] format (MPEG-G) | [Under development] | [Under development] | Context-adaptive binary arithmetic coding (CABAC) | https://github.com/mitogen/genie | BSD | [21] |
| Software | Description | Compression Ratio | Data Used for Evaluation | Approach/Encoding Scheme | Link | Use License | Reference |
|---|---|---|---|---|---|---|---|
| Genome Differential Compressor (GDC) | LZ77-style tool for compressing multiple genomes of the same species | 180 to 250-fold / 70 to 100-fold | Nuclear genome sequence of human and Saccharomyces cerevisiae | Huffman coding | http://sun.aei.polsl.pl/gdc | GPLv2 | [5] |
| Genome Re-Sequencing (GRS) | Reference sequence-based tool independent of a reference SNP map or sequence variation information | 159-fold / 18,133-fold / 82-fold | Nuclear genome sequence of human, Arabidopsis thaliana (different revisions of the same genome), and Oryza sativa | Huffman coding | https://web.archive.org/web/20121209070434/http://gmdd.shgmo.org/Computational-Biology/GRS/ | free of charge for non-commercial use | [6] |
| Genome Re-sequencing Encoding (GReEN) | Probabilistic copy model-based tool for compressing re-sequencing data using a reference sequence | ~100-fold | Human nuclear genome sequence | Arithmetic coding | http://bioinformatics.ua.pt/software/green/ | -Undeclared- | [7] |
| DNAzip | A package of compression tools | ~750-fold | Human nuclear genome sequence | Huffman coding | http://www.ics.uci.edu/~dnazip/ | -Undeclared- | [9] |
| GenomeZip | Compression with respect to a reference genome. Optionally uses external databases of genomic variations (e.g. dbSNP) | ~1200-fold | Human nuclear genome sequence (Watson) and sequences from the 1000 Genomes Project | Entropy coding for approximations of empirical distributions | https://sourceforge.net/projects/genomezip/ | -Undeclared- | [10] |
References
- Giancarlo, R., D. Scaturro, and F. Utro. 2009. Textual data compression in computational biology: a synopsis. Bioinformatics 25(13): 1575-1586.
- Nalbantoglu, Ö. U., D. J. Russell, and K. Sayood. 2010. Data compression concepts and algorithms and their applications to bioinformatics. Entropy 12(1): 34-52.
- Hosseini, D., Pratas, and A. Pinho. 2016. A survey on data compression methods for biological sequences. Information 7(4):(2016): 56
- Brandon, M. C., D. C. Wallace, and P. Baldi. 2009. Data structures and compression algorithms for genomic sequence data. Bioinformatics 25(14): 1731–1738.
- Deorowicz, S., and S. Grabowski. 2011. Robust relative compression of genomes with random access. Bioinformatics 27(21): 2979-2986.
- Wang, C., and D. Zhang. 2011. A novel compression tool for efficient storage of genome resequencing data. Nucleic Acids Res 39(7): e45.
- Pinho, A. J., D. Pratas, and S. P. Garcia. 2012. GReEn: a tool for efficient compression of genome resequencing data. Nucleic Acids Res 40(4): e27.
- Tembe, W., J. Lowey, and E. Suh. 2010. G-SQZ: Compact encoding of genomic sequence and quality data. Bioinformatics 26(17): 2192-2194.
- Christley, S., Y. Lu, C. Li, and X. Xie. 2009. Human genomics as email attachments. Bioinformatics 25(2): 274-275.
- Pavlichin, D.S., Weissman, T., and G. Yona. 2013. The human genome contracts again. Bioinformatics 29(17): 2199-2202.
- Kuruppu, S., S. J. Puglisi, and J. Zobel. 2011. Reference sequence construction for relative compression of genomes. Lecture Notes in Computer Science 7024: 420-425.
- Grabowski, S., and S. Deorowicz. 2011. Engineering Relative Compression of Genomes. In Proceedings of CoRR.
- Pratas, D., Pinho, A. J., and Ferreira, P. J. S. G. Efficient compression of genomic sequences. Data Compression Conference, Snowbird, Utah, 2016.
- Lan, D., et al. 2021 Genozip: a universal extensible genomic data compressor, Bioinformatics https://doi.org/10.1093/bioinformatics/btab102
- CRAM benchmarking http://www.htslib.org/benchmarks/CRAM.html
- CRAM format specification (version 3.0) https://samtools.github.io/hts-specs/CRAMv3.pdf
- "The Importance of Data Compression in the Field of Genomics" (in en-US). https://pulse.embs.org/march-2019/the-importance-of-data-compression-in-the-field-of-genomics/.
- "ISO/IEC 23092-2:2019 Information technology — Genomic information representation — Part 2: Coding of genomic information" (in en-US). https://www.iso.org/standard/73536.html.
- "An introduction to MPEG-G, the new ISO standard for genomic information representation". https://www.biorxiv.org/content/10.1101/426353v1/.
- "ISO/IEC 23092-2:2019 Information technology — Genomic information representation — Part 2: Coding of genomic information" (in en-US). https://www.iso.org/standard/73536.html.
- "An introduction to MPEG-G, the new ISO standard for genomic information representation". https://www.biorxiv.org/content/10.1101/426353v2/.

