 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jaqueline Loaeza | + 1120 word(s) | 1120 | 2020-11-20 06:55:12 | | | |
| 2 | Lily Guo | -48 word(s) | 1072 | 2020-12-03 04:55:32 | | |
Video Upload Options
The human genome has additional regulation layers for the regulation of transcription. The DNA methylation is a key epigenetic process that sharp in functional regions in the genome. The 5-methylcytosine patterns in promoter regions are related to gene expression regulation.
1. Introduction
DNA methylation is one of the best-characterized epigenetic modifications, and it has a critical role in active and inactive chromatin equilibrium for gene expression control[1]. Gene silencing by DNA methylation is necessary to balance and regulate cell biological processes. DNA methylation is a chemical modification that occurs in the carbon 5 of cytosines located at the 5′ of guanine, and this arrangement is called CpG dinucleotide[2]. DNA methylation augments the information contained in the DNA sequence, and confers read duality to the same sequence, changing the functional status of a gene from active to inactive or vice versa[3][1]. DNA methylation is deposited on a CpG context by DNA methyltransferases (DNMTs). DNMTs are a family of mammalian enzymes that convert cytosine to 5-methylcytosine (5mC). In humans, there are three catalytically active DNMTs: DNMT1, DNMT3A, and DNMT3B. DNMT1 is responsible for maintaining existing methylation, while DNMT3A and DNMT3B establish dynamic methylation patterns. These enzymes are finely regulated, and deregulation of their expression leads to abnormal methylation[4][5]. The strict regulation of genes via methylation ranges from specific allele methylation patterns to differentially expressed genes between stem cells and adult cells. Behind the DNMTs activity and DNA methylation deposition, there are key mechanisms through cis- and trans-regulatory factors that influence DNA methylation. Cis-regulatory elements (CREs) form a key class of regulatory noncoding DNA sequences, which act to regulate transcription of a neighboring gene. Structurally, the genome is segmented into biologically active cis elements, such as enhancers, insulators, locus control region, promoters, CpG islands (CGIs), and binding sites for transcription factors[6], which can be methylated as a second regulatory level[7] [8]. Moreover, trans-regulatory proteins act through two domains, first by binding to a particular DNA domain (Zn fingers, Leu zipper), and the second is responsible for activity on transcription (as rich in Gln, rich in Pro, acidic α-helix domain)[8][9]. In the human genome, DNA methylation is spread in around 28 million CpG dinucleotides distributed in more than half of the genes and can be directed by cis and trans elements. CpG dinucleotides are commonly condensed in regions called CpG islands that are short interspersed DNA sequences with an average size of 500 to 1000 bp and >60% CpG content relative to the bulk genome[10]. CGIs are frequently found in the regulatory region of about 72%[11] of protein-coding genes and are known to impact their transcriptional regulation by modulating the accessibility and affinity of transcription factors to their binding sites[12][13]. The promoter regions and CGIs from methylated genes also contain smaller common sequences that are short DNA motifs. These sequences can promote the affinity of DNMTs by an unknown mechanism. These motifs are found in DNMTs’ enrichment peak and suggest that they can be binding motifs for proteins or other molecules that regulate DNA methylation[14] [15]. The DNA methylation landscape is molded by other epigenetics modifiers such as long noncoding RNAs (lncRNAs) and histone modifications. LncRNAs are RNAs with a length of more than 200 nucleotides and do not encode proteins but can interact with DNMTs and localize them to specific genes[15][16]. Histone modifications contribute to the regulation of gene expression and are associated with DNMTs´ function in several genes. Histones are central components of chromatin, and the residues of histone tail may undergo post-translational modifications such as phosphorylation, acetylation, and methylation. The complex dynamic combination of these histone marks regulates active or repressive chromatin states. In particular, mono-, di-, and trimethylation of lysines is associated with positive or negative chromatin methylation landscapes[17][18].
2. Discovery of 5-Methylcytosine and Its Function in the Genome
In vitro, in early studies on the composition and biochemical properties of nucleic acids, 5mC was discovered, and it was described as an unknown element in the DNA[19]. Later, while differences between pathogenic and nonpathogenic bacteria were being searched for, 5mC was confirmed as a product of the hydrolysis of Bacillus tuberculosis nucleic acids and was identified as the fourth pyrimidine (cytosine, thymine, uracil, and 5-methylcytosine)[19][20]. Consequently, 5mC was identified in mammalian DNA and was described with acid–alkaline properties similar to cytosine, but without being uracil[21]. Almost parallel to the reports of 5mC, in 1939, Waddington proposed the existence of an epigenotype to explain certain aspects of development that were influenced by the environment and not the result of changes in the genotype[22]. Functional studies of DNA methylation evolved from a protective mechanism against foreign DNA in bacteria to a regulatory mechanism of gene expression in vertebrates[23]. Studies in Escherichia coli demonstrated that when foreign DNA was not digested by endonucleases, the DNA was methylated as an alternative host protection mechanism[24]. Based on prokaryotic observations regarding the presence of enzymes that methylate DNA and after visualization of problems with the existent models, Rigss et al. intuited the existence of mammals’ DNMTs and consequently raised the X-chromosome inactivation model via DNA methylation[25] [26]. From studies in both vertebrates and plants[26]emerged the first hint of 5mC in gene regulation in 1975[27] [28]. The direct correlation between gene repression and differentiation was established in 1980. Globin genes were analyzed by means of restriction endonucleases, which are unable to cleave methylated DNA (HpaII). In the germ line, all sites tested in the globin gene region were methylated; however, in somatic tissue, DNA methylation was absent at specific sites in the globin gene[28]. Additionally, the treatment with Cytidine analog (5-azacytidine) in mouse embryo cells induced changes in the differentiation as a consequence of methylation inhibition of newly synthesized DNA[29] . At this point, the aforementioned assays confirmed the existence of specific methylation patterns, symmetric in both chains, heritable, and tissue-specific[28]. Gruenbaum and Bestor and Ingram worked on the pioneering studies of vertebrate DNMTs. These studies included substrate-dependent DNA methylation that showed DNMTs preference for hemimethylated CpG sequences, and that later was established as their methylation mechanism and function[4][27]. DNMTs were sequenced and cloned from mouse cells to study their functional domains and decipher their action mechanism[28]. DNMTs and DNA methylation was established as an integral component of the gene expression regulation in mammals (Figure 1). The global DNA methylation patterns in mammals are established by three enzymatically active DNA-methyltransferases: DNMT1, DNMT3A, and DNMT3B.
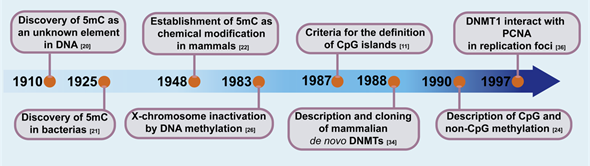
Figure 1. Timeline of key findings that led to the discovery and role definition DNA methyltransferases (DNMTs) in mammalian genomes and gene expression.
References
- Lee, H.J.; Hore, T.A.; Reik, W. Reprogramming the Methylome: Erasing Memory and Creating Diversity. Cell Stem Cell 2014, 14, 710–719, doi:10.1016/j.stem.2014.05.008.
- Gruenbaum, Y.; Cedar, H.; Razin, A. Substrate and sequence specificity of a eukaryotic DNA methylase. Nat. Cell Biol. 1982, 295, 620–622, doi:10.1038/295620a0.
- Schübeler, D. Function and information content of DNA methylation. Nat. Cell Biol. 2015, 517, 321–326, doi:10.1038/nature14192.
- Rhee, I.; Bachman, K.E.; Park, B.H.; Jair, K.-W.; Yen, R.-W.C.; Schuebel, K.E.; Cui, H.; Feinberg, A.P.; Lengauer, C.; Kinzler, K.W.; et al. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nat. Cell Biol. 2002, 416, 552–556, doi:10.1038/416552a.
- Bestor, T.H. The DNA methyltransferases of mammals. Hum. Mol. Genet. 2000, 9, 2395–2402, doi:10.1093/hmg/9.16.2395.
- Riethoven, J.-J.M. Regulatory Regions in DNA: Promoters, Enhancers, Silencers, and Insulators. Methods Mol. Biol. 2010, 674, 33–42, doi:10.1007/978-1-60761-854-6_3.
- Angeloni, A.; Bogdanovic, O. Enhancer DNA methylation: Implications for gene regulation. Essays Biochem. 2019, 63, 707–715, doi:10.1042/ebc20190030.
- Wittkopp, P.J.; Kalay, G. Cis-regulatory elements: Molecular mechanisms and evolutionary processes underlying divergence. Nat. Rev. Genet. 2011, 13, 59–69, doi:10.1038/nrg3095.
- Shibata, M.; Gulden, F.O.; Sestan, N. From trans to cis: Transcriptional regulatory networks in neocortical development. Trends Genet. 2015, 31, 77–87, doi:10.1016/j.tig.2014.12.004.
- Gardiner-Garden, M.; Frommer, M. CpG Islands in vertebrate genomes. J. Mol. Biol. 1987, 196, 261–282, doi:10.1016/0022-2836(87)90689-9.
- Saxonov, S.; Berg, P.; Brutlag, D.L. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. USA 2006, 103, 1412–1417, doi:10.1073/pnas.0510310103.
- Lienert, F.; Wirbelauer, C.; Som, I.; Dean, A.; Mohn, F.; Schübeler, D. Identification of genetic elements that autonomously determine DNA methylation states. Nat. Genet. 2011, 43, 1091–1097, doi:10.1038/ng.946.
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022, doi:10.1101/gad.2037511.
- Takahashi, M.; Kamei, Y.; Ehara, T.; Yuan, X.; Suganami, T.; Takai-Igarashi, T.; Hatada, I.; Ogawa, Y. Analysis of DNA methylation change induced by Dnmt3b in mouse hepatocytes. Biochem. Biophys. Res. Commun. 2013, 434, 873–878, doi:10.1016/j.bbrc.2013.04.041.
- Wang, L.; Zhao, Y.; Bao, X.; Zhu, X.; Kwok, Y.K.-Y.; Sun, K.; Chen, X.; Huang, Y.; Jauch, R.; Esteban, M.A.; et al. LncRNA Dum interacts with Dnmts to regulate Dppa2 expression during myogenic differentiation and muscle regeneration. Cell Res. 2015, 25, 335–350, doi:10.1038/cr.2015.21.
- Li, Y.; Cheng, C. Long noncoding RNA NEAT1 promotes the metastasis of osteosarcoma via interaction with the G9a-DNMT1-Snail complex. Am. J. Cancer Res. 2018, 8, 81–90.
- Schlesinger, Y.; Straussman, R.; Keshet, I.; Farkash, S.; Hecht, M.; Zimmerman, J.; Eden, E.; Yakhini, Z.; Ben-Shushan, E.; Reubinoff, B.E.; et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat. Genet. 2006, 39, 232–236, doi:10.1038/ng1950.
- Dhayalan, A.; Rajavelu, A.; Rathert, P.; Tamas, R.; Jurkowska, R.Z.; Ragozin, S.; Jeltsch, A. The Dnmt3a PWWP Domain Reads Histone 3 Lysine 36 Trimethylation and Guides DNA Methylation. J. Biol. Chem. 2010, 285, 26114–26120, doi:10.1074/jbc.m109.089433.
- Hotchkiss, R.D. The quantitative separation of purines, pyrimidines, and nucleosides by paper chromatography. J. Biol. Chem. 1948, 175, 315–322.
- Just, T.; Waddington, C.H. An Introduction to Modern Genetics. Am. Midl. Nat. 1939, 22, 754, doi:10.2307/2420355.
- Bestor, T.H. DNA methylation: Evolution of a bacterial immune function into a regulator of gene expression and genome structure in higher eukaryotes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1990, 326, 179–187.
- Dugaiczyk, A.; Hedgpeth, J.; Boyer, H.W.; Goodman, H.M. Physical identity of the SV40 deoxyribonucleic acid sequence recognized by the Eco RI restriction endonuclease and modification methylase. Biochemistry 1974, 13, 503–512, doi:10.1021/bi00700a016.
- Riggs, A. X inactivation, differentiation, and DNA methylation. Cytogenet. Genome Res. 1975, 14, 9–25, doi:10.1159/000130315.
- Vanyushin, B.F.; Tkacheva, S.G.; Belozersky, A.N. Rare Bases in Animal DNA. Nat. Cell Biol. 1970, 225, 948–949, doi:10.1038/225948a0.
- Holliday, R.; Pugh, J. DNA modification mechanisms and gene activity during development. Science 1975, 187, 226–232, doi:10.1126/science.1111098.
- Van Der Ploeg, L.H.; Flavell, R.A. DNA methylation in the human γ delta β -globin locus in erythroid and nonerythroid tissues. Cell 1980, 19, 947–958.
- Jones, P.A.; Taylor, S.M. Cellular differentiation, cytidine analogs and DNA methylation. Cell 1980, 20, 85–93, doi:10.1016/0092-8674(80)90237-8.
- Razin, A.; Riggs, A.D. DNA methylation and gene function. Science 1980, 210, 604–610, doi:10.1126/science.6254144.
- Razin, A.; Riggs, A.D. DNA methylation and gene function. Science 1980, 210, 604–610, doi:10.1126/science.6254144.
- Johnson, T.B.; Coghill, R.D. researches on pyrimidines. C111. The discovery of 5-methyl-cytosine in tuberculinic acid, the nucleic acid of the tubercle bacillus. J. Am. Chem. Soc. 1925, 47, 2838–2844, doi:10.1021/ja01688a030.

