 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | mina Student ardestani | -- | 3066 | 2022-11-06 07:42:34 | | | |
| 2 | Rita Xu | Meta information modification | 3066 | 2022-11-07 03:07:42 | | |
Video Upload Options
Heat shock proteins (Hsps) have garnered special attention in cancer therapy as molecular chaperones with regulatory/mediatory effects on folding, maintenance/stability, maturation, and conformation of proteins as well as their effects on prevention of protein aggregation. Hsp90 ensures the stability of various client proteins needed for the growth of cells or the survival of tumor cells; therefore, they are overexpressed in tumor cells and play key roles in carcinogenesis. Accordingly, Hsp90 inhibitors are recognized as attractive therapeutic agents for investigations pertaining to tumor suppression. Natural Hsp90 inhibitors comprising geldanamycin (GM), reclaimed analogs of GM including 17-AAG and DMAG, and radicicol, a natural macrocyclic antifungal, are among the first potent Hsp90 inhibitors.
1. Introduction
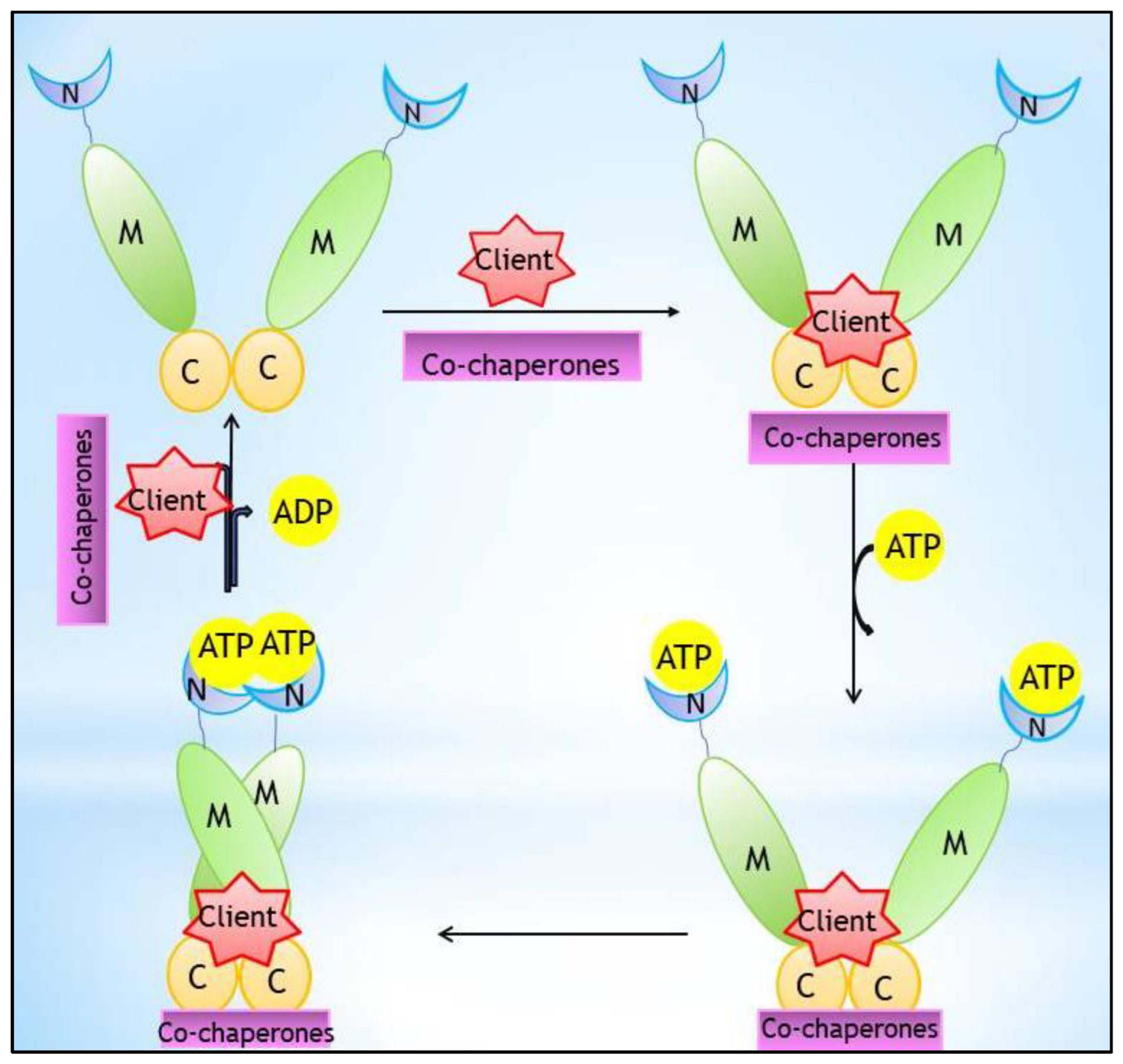
2. Hsp90 Inhibitors
2.1. Natural Inhibitors
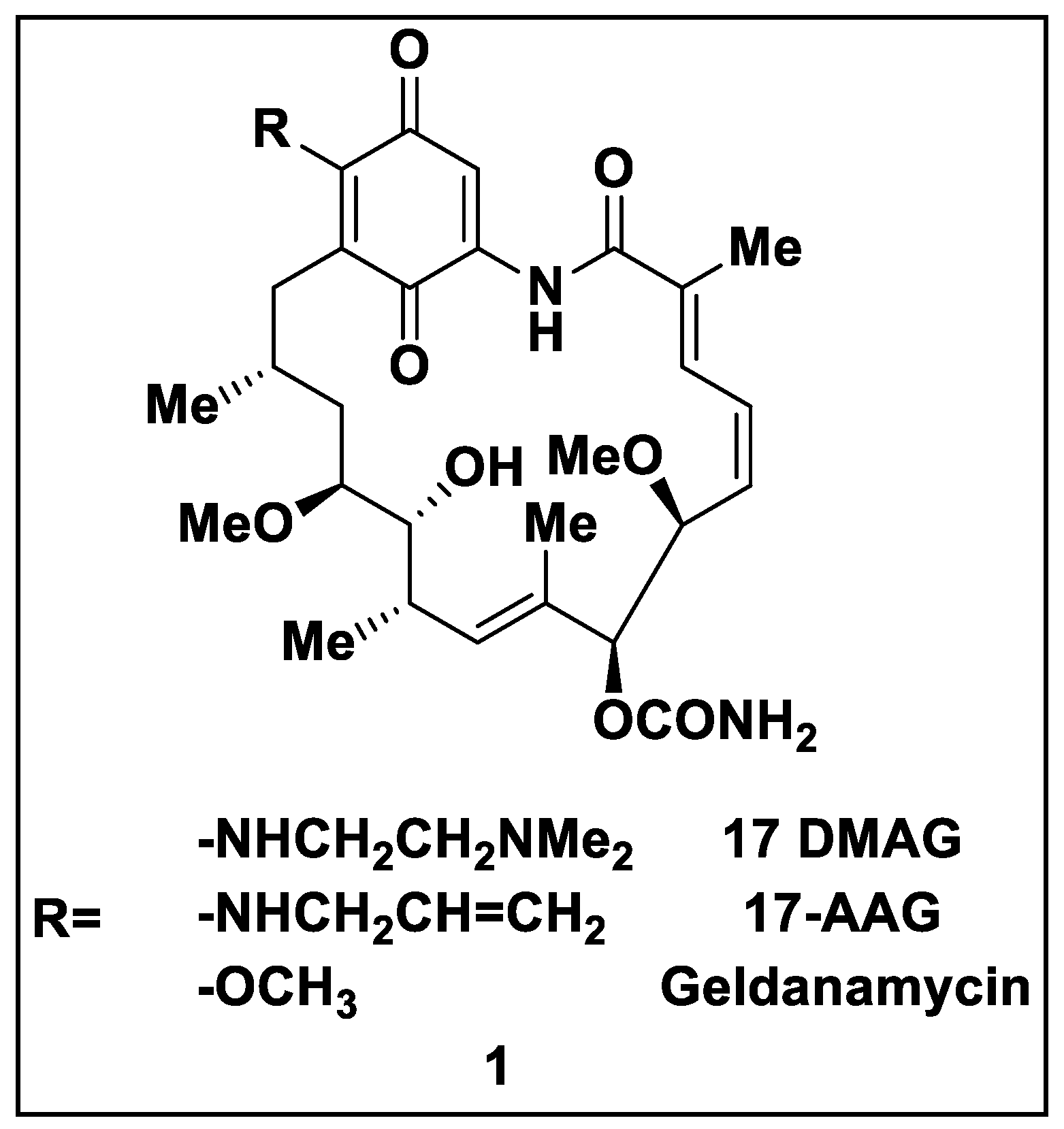
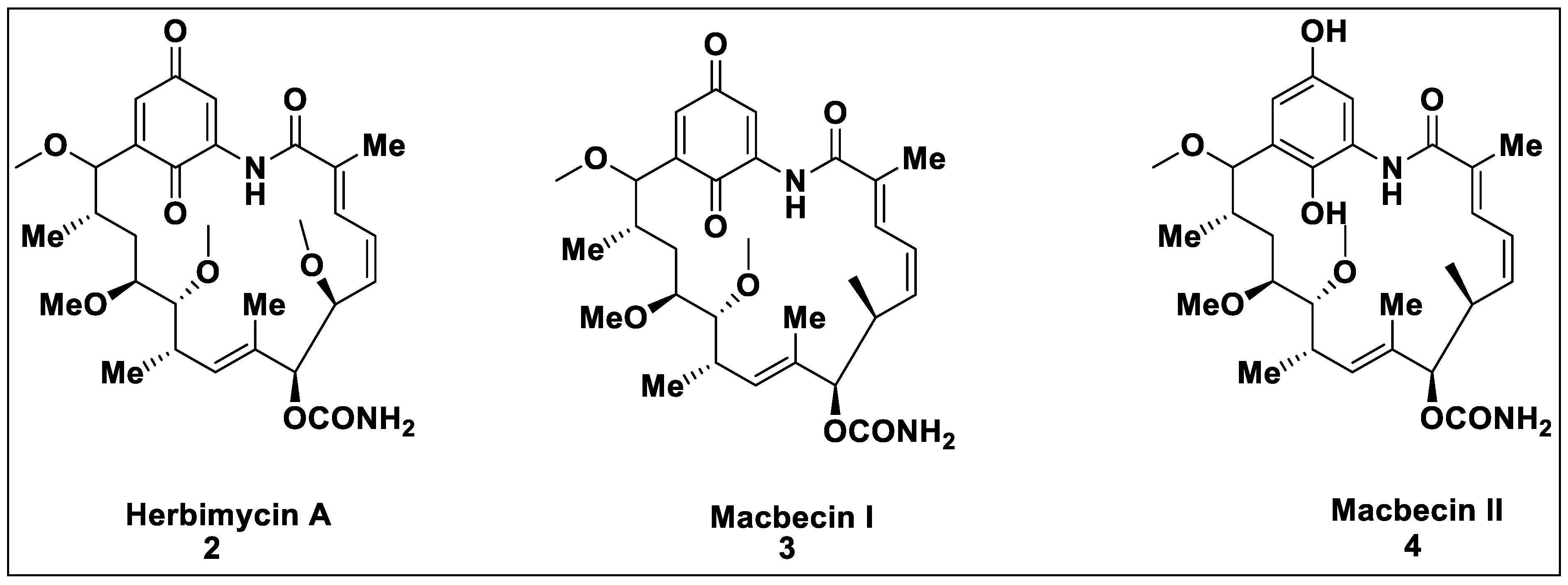
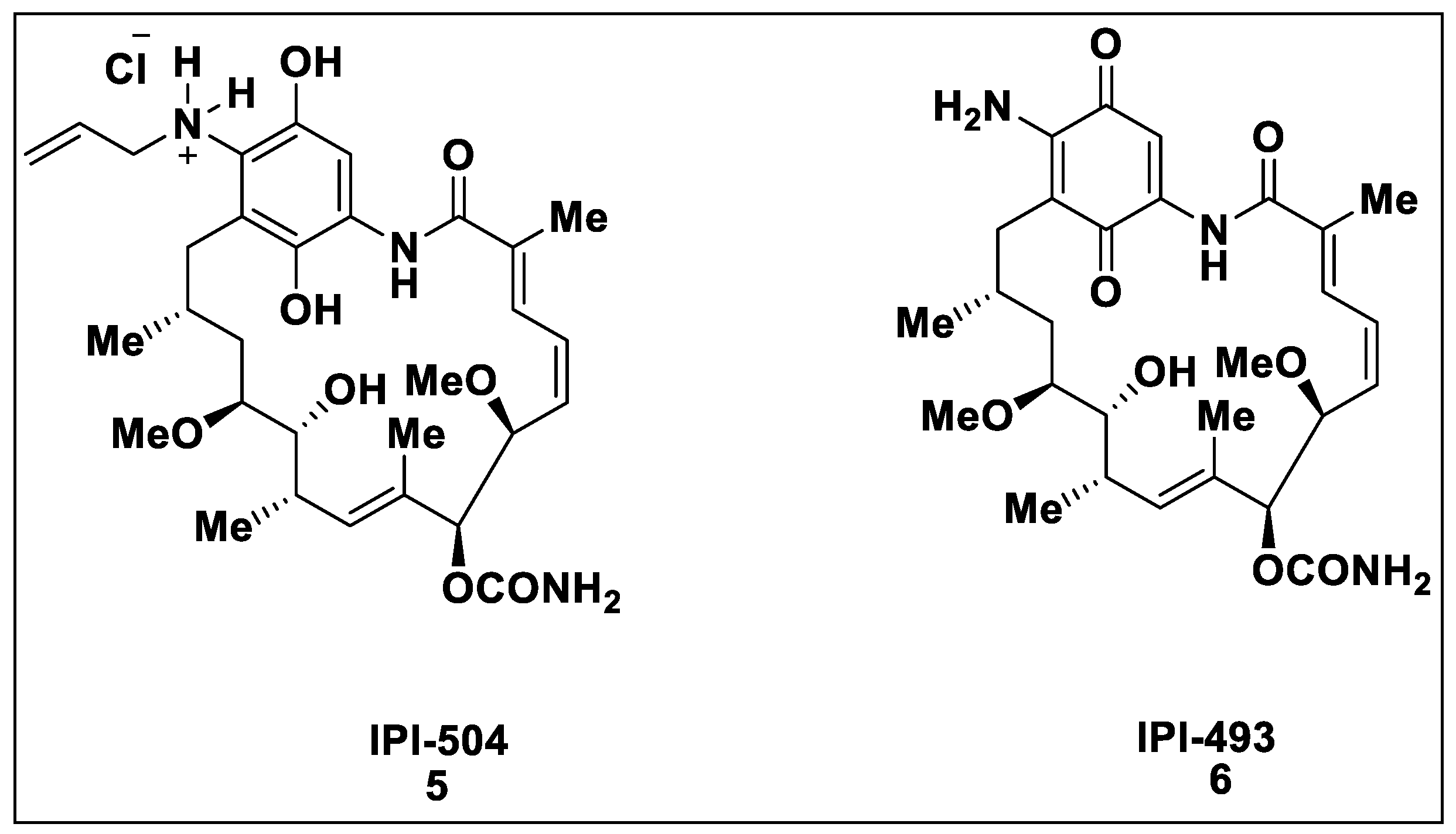
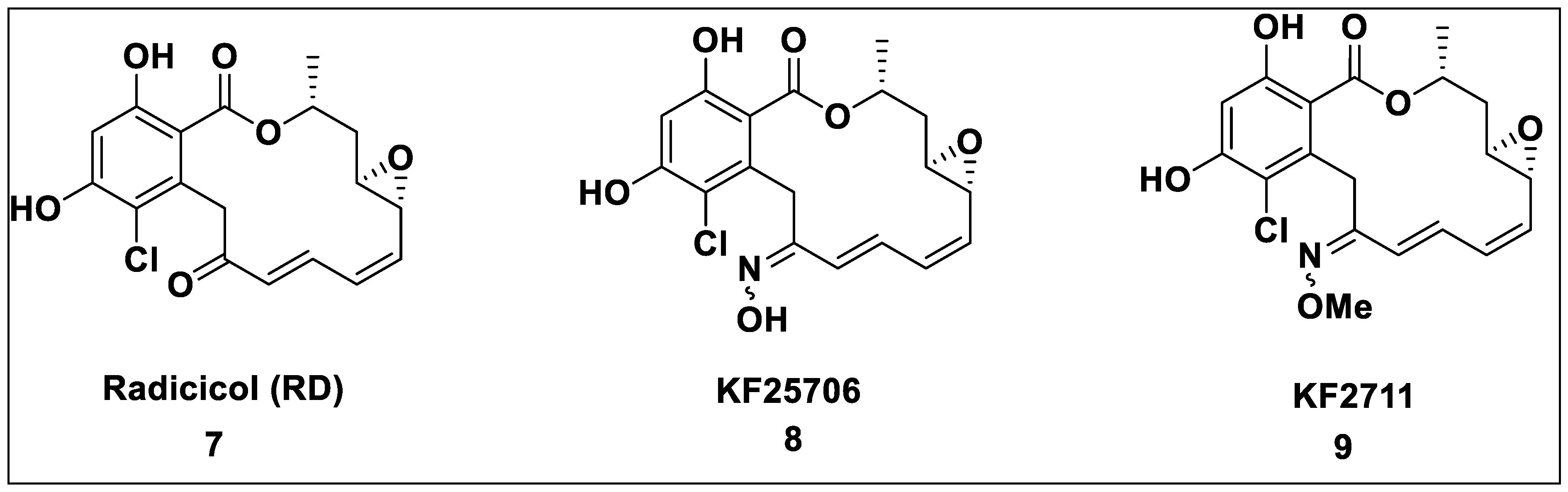
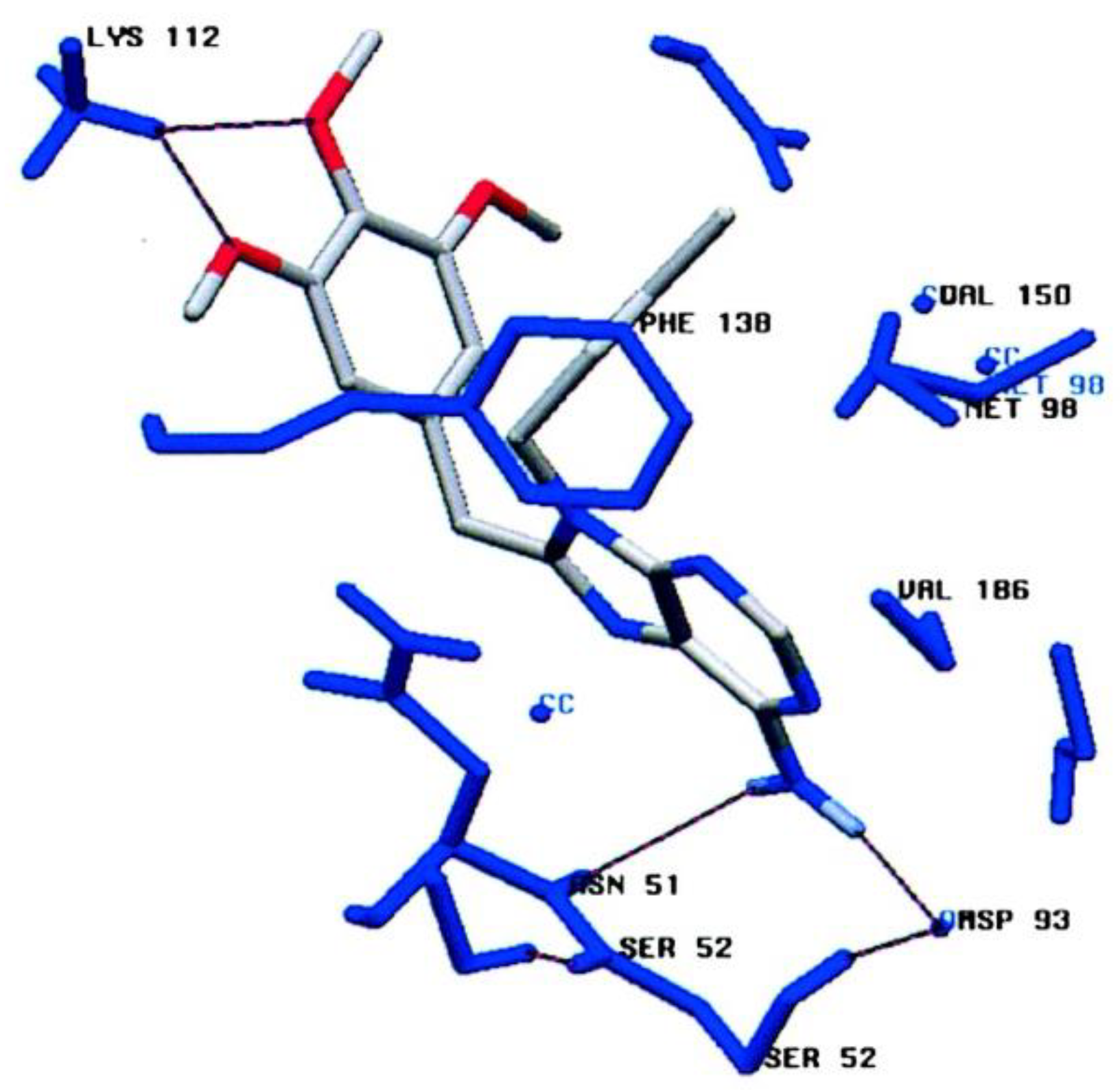
2.2. Synthetic Hsp90 Inhibitors
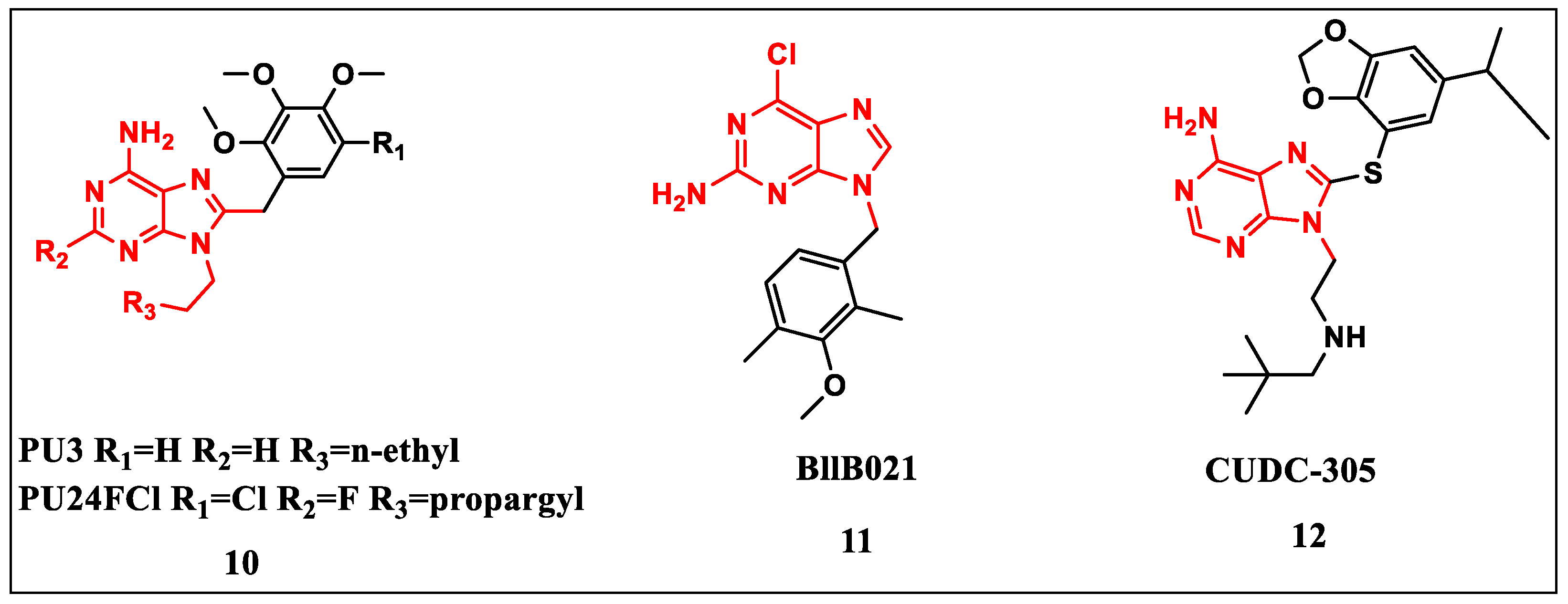
2.2.1. Purine-Based Structures
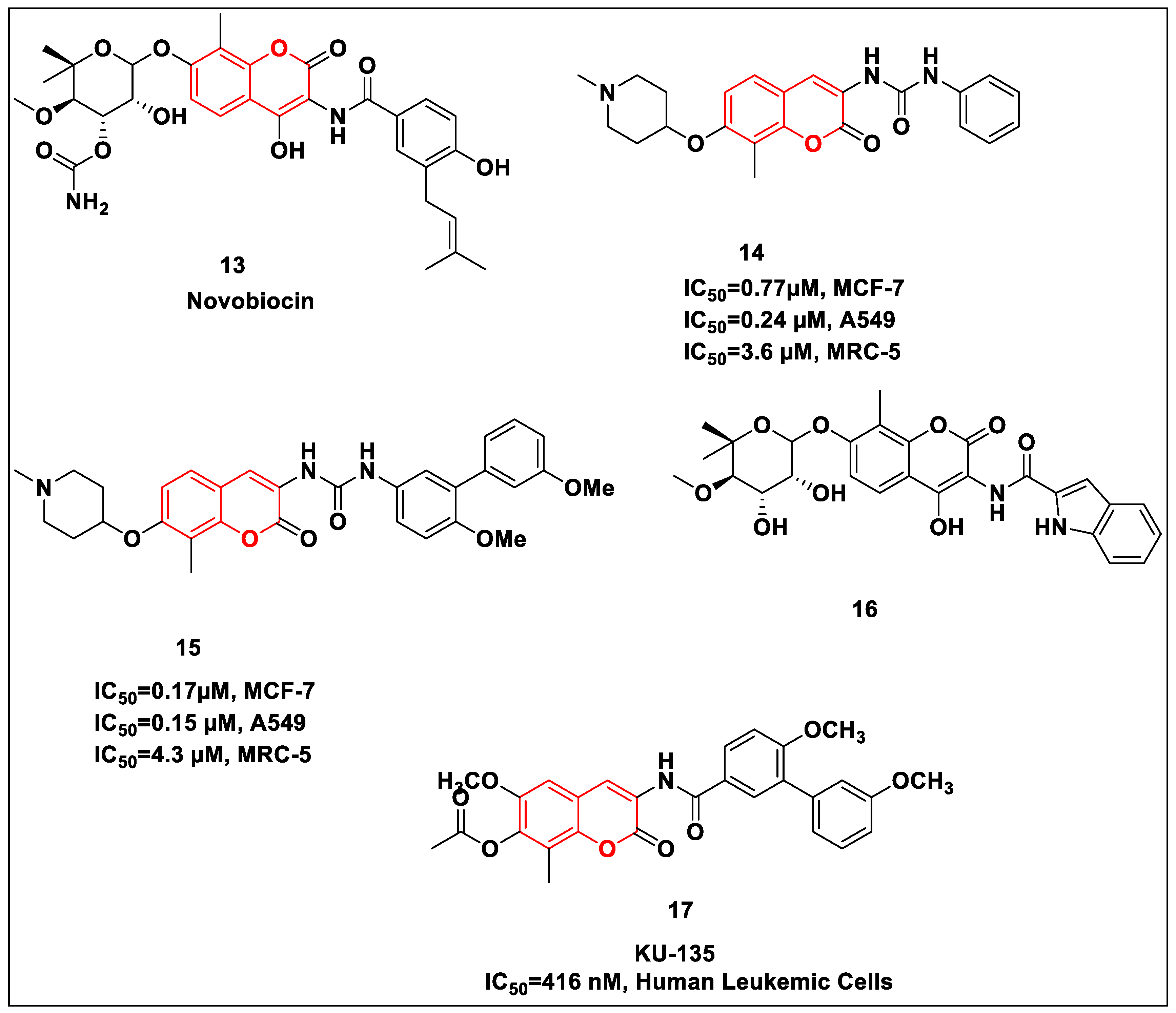
2.2.2. Coumarin-Based Structures
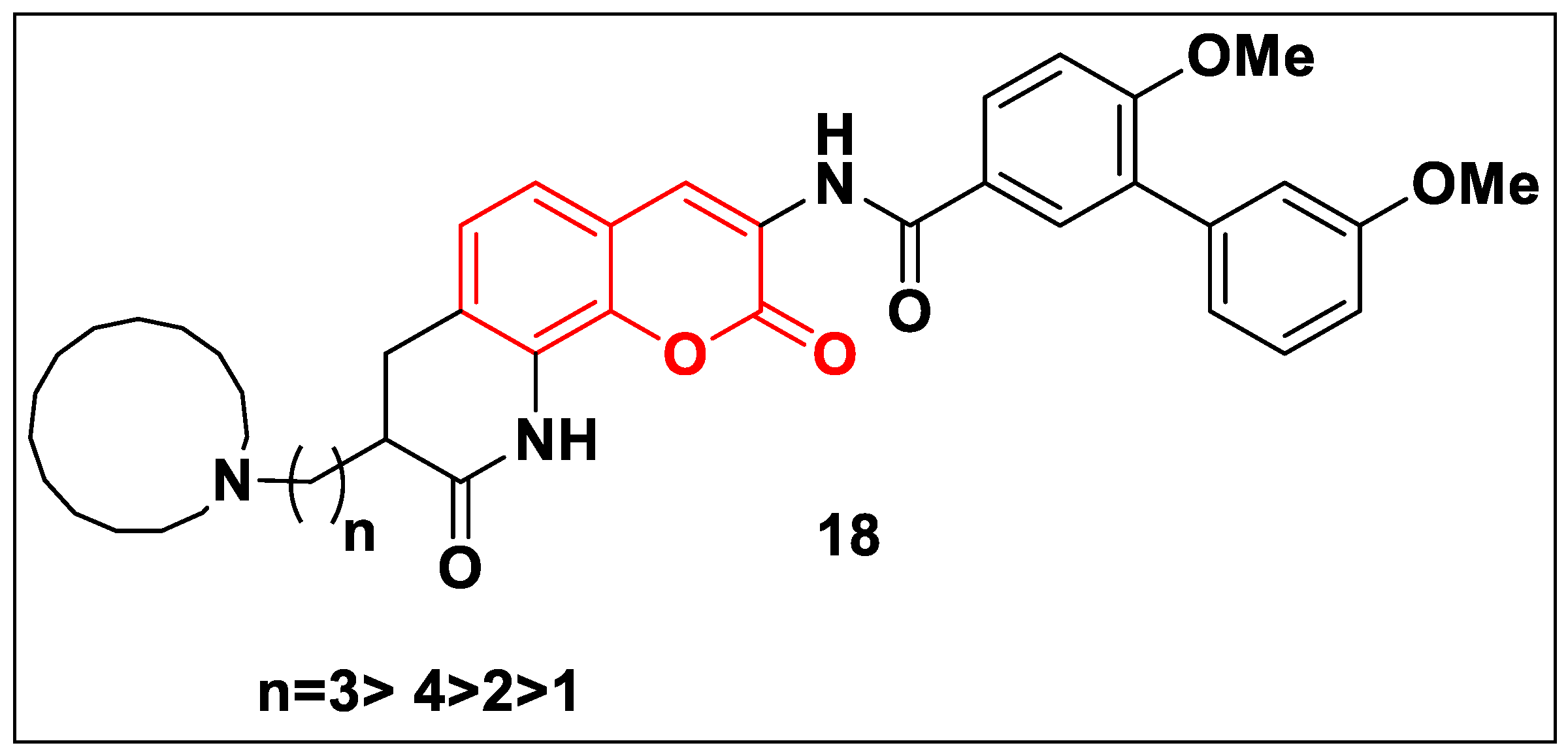
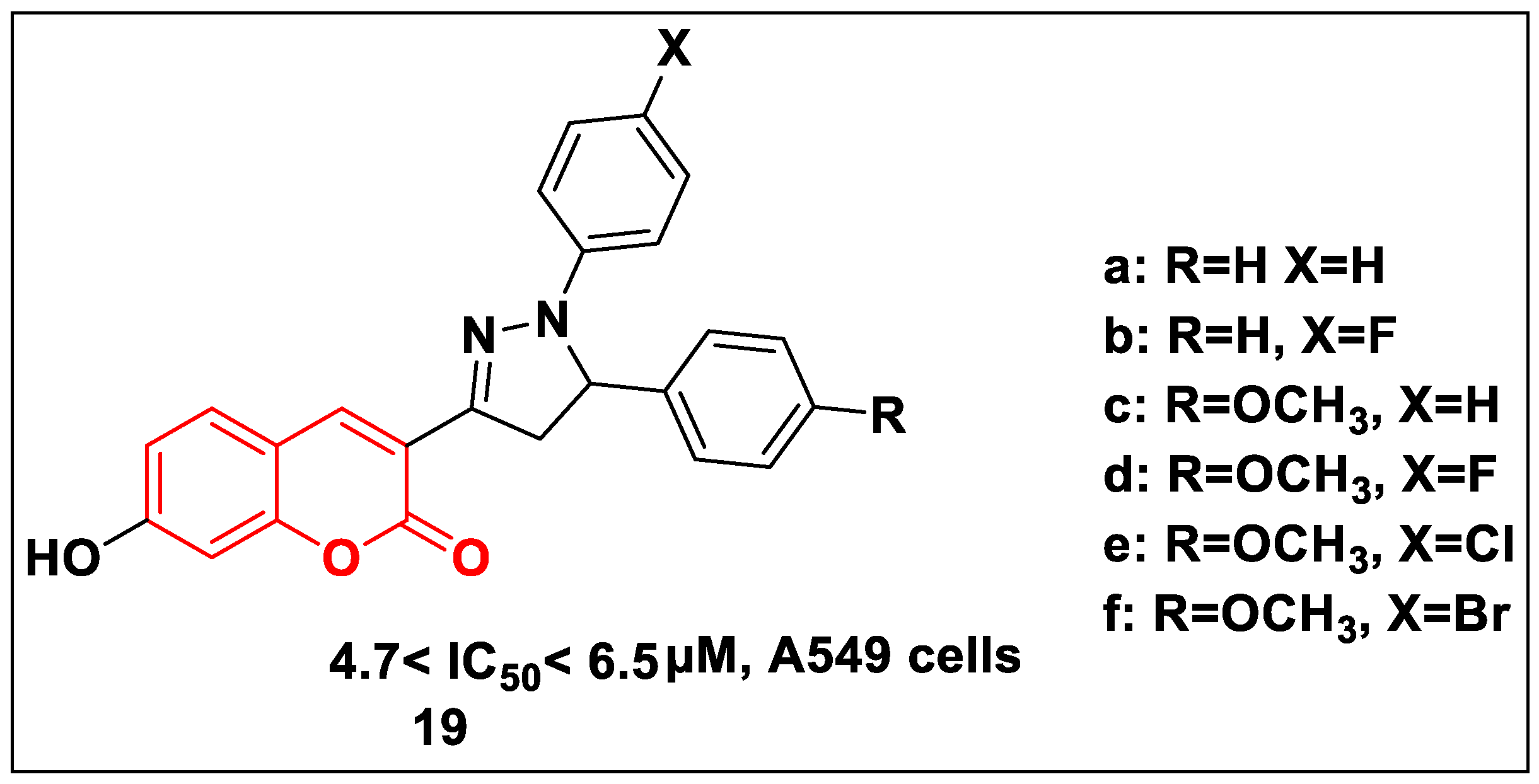
2.2.3. Quinolone-Based Structures
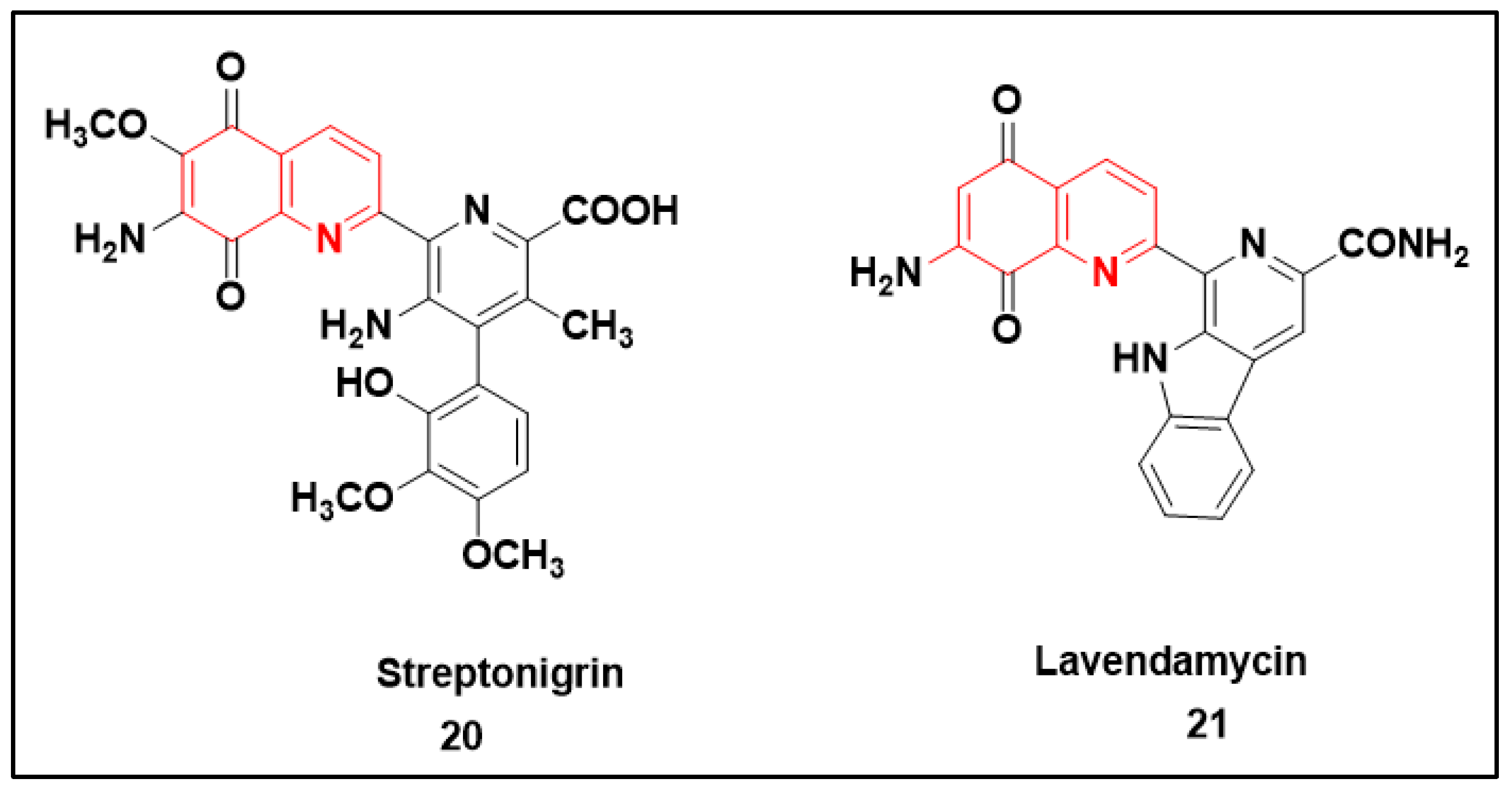
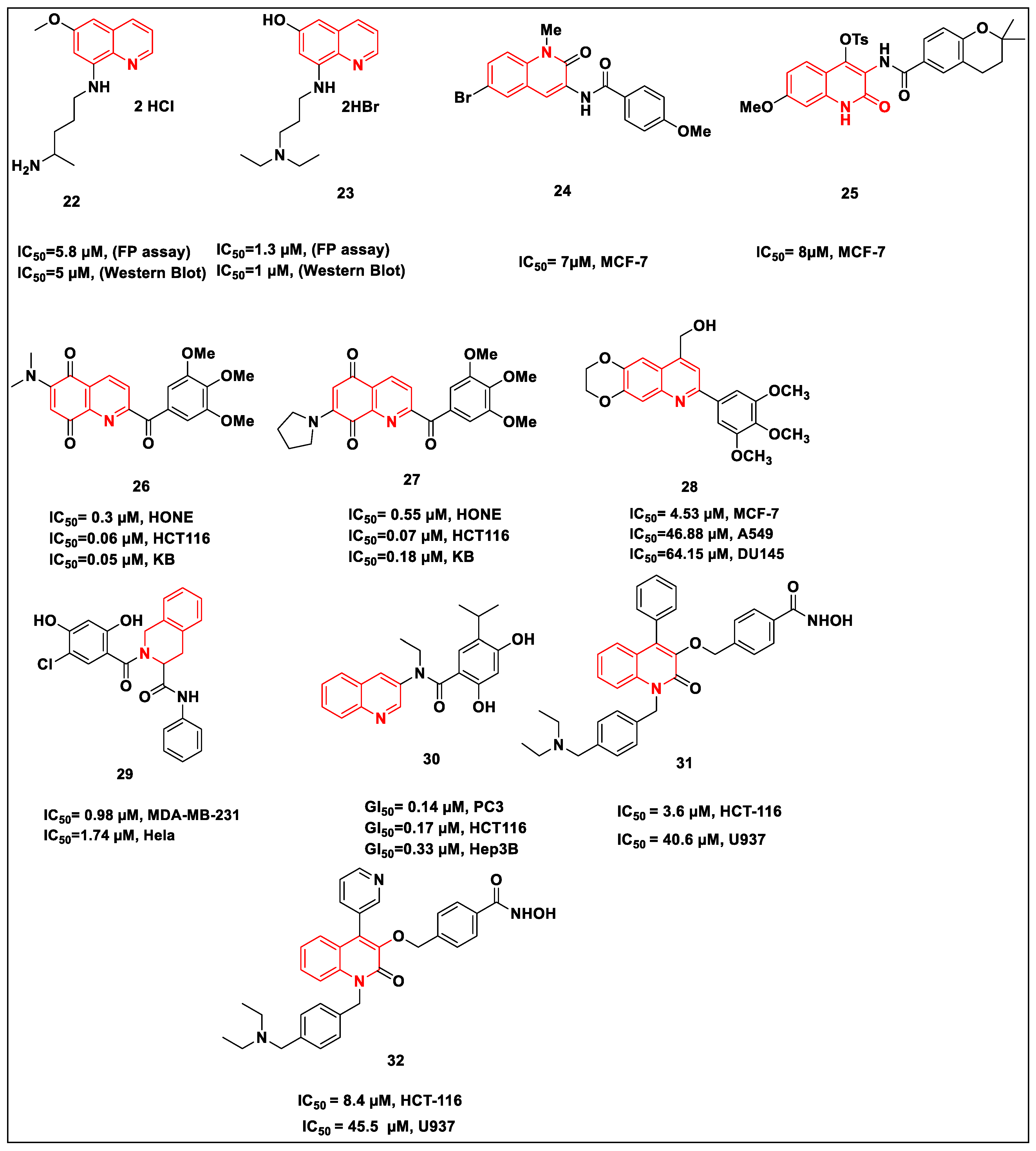
References
- Mahboubi-Rabbani, M.; Zarghi, A. Lipoxygenase Inhibitors as Cancer Chemopreventives: Discovery, Recent Developments and Future Perspectives. Curr. Med. Chem. 2021, 28, 1143–1175.
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The Hsp90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360.
- Mahalingam, D.; Swords, R.; Carew, J.S.; Nawrocki, S.T.; Bhalla, K.; Giles, F.J. Targeting HSP90 for cancer therapy. Br. J. Cancer 2009, 100, 1523–1529.
- Ischia, J.; So, A.I. The role of heat shock proteins in bladder cancer. Nat. Rev. Urol. 2013, 10, 386–395.
- Davenport, J.; Manjarrez, J.R.; Peterson, L.; Krumm, B.; Blagg, B.S.J.; Matts, R.L. Gambogic Acid, a Natural Product Inhibitor of Hsp90. J. Nat. Prod. 2011, 74, 1085–1092.
- Bhat, R.; Tummalapalli, S.R.; Rotella, D.P.J. Progress in the Discovery and Development of Heat Shock Protein 90 (Hsp90) Inhibitors: Miniperspective. Med. Chem. 2014, 57, 8718–8728.
- Jaeger, A.M.; Whitesell, L. Hsp90: Enabler of Cancer Adaptation. Annu. Rev. Cancer Biol. 2019, 3, 275–297.
- Gupta, S.D.; Bommaka, M.K.; Banerjee, A. Inhibiting protein-protein interactions of Hsp90 as a novel approach for targeting cancer. Eur. J. Med. Chem. 2019, 178, 48–63.
- Wang, L.; Zhang, Q.; You, Q. Targeting the HSP90–CDC37–kinase chaperone cycle: A promising therapeutic strategy for cancer. Med. Res. Rev. 2022, 42, 156–182.
- Li, L.; Chen, N.-N.; You, Q.-D.; Xu, X.-L. An updated patent review of anticancer Hsp90 inhibitors (2013–present). Expert Opin. Ther. Patents 2021, 31, 67–80.
- Niu, M.; Song, S.; Su, Z.; Wei, L.; Li, L.; Pu, W.; Zhao, C.; Ding, Y.; Wang, J.; Cao, W.; et al. Inhibition of heat shock protein (HSP) 90 reverses signal transducer and activator of transcription (STAT) 3-mediated muscle wasting in cancer cachexia mice. Br. J. Pharmacol. 2021, 178, 4485–4500.
- Jafari, A.; Rezaei-Tavirani, M.; Farhadihosseinabadi, B.; Taranejoo, S.; Zali, H. HSP90 and Co-chaperones: Impact on Tumor Progression and Prospects for Molecular-Targeted Cancer Therapy. Cancer Investig. 2020, 38, 310–328.
- Sanchez, J.; Carter, T.R.; Cohen, M.S.; Blagg, B.S.J. Old and New Approaches to Target the Hsp90 Chaperone. Curr. Cancer Drug Targets 2020, 20, 253–270.
- Trepel, J.; Mollapour, M.; Giaccone, G.; Neckers, L. Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 2010, 10, 537–549.
- Nazaripour, E.; Mousazadeh, F.; Moghadam, M.D.; Najafi, K.; Borhani, F.; Sarani, M.; Ghasemi, M.; Rahdar, A.; Iravani, S.; Khatami, M. Biosynthesis of lead oxide and cerium oxide nanoparticles and their cytotoxic activities against colon cancer cell line. Inorg. Chem. Commun. 2021, 131, 108800.
- Iravani, S.; Varma, R.S. MXenes for Cancer Therapy and Diagnosis: Recent Advances and Current Challenges. ACS Biomater. Sci. Eng. 2021, 7, 1900–1913.
- Delfi, M.; Sartorius, R.; Ashrafizadeh, M.; Sharifi, E.; Zhang, Y.; De Berardinis, P.; Zarrabi, A.; Varma, R.S.; Tay, F.R.; Smith, B.R.; et al. Self-assembled peptide and protein nanostructures for anti-cancer therapy: Targeted delivery, stimuli-responsive devices and immunotherapy. Nano Today 2021, 38, 101119.
- Alavi, M.; Varma, R.S. Overview of Novel Strategies for the Delivery of Anthracyclines to Cancer Cells by Liposomal and Polymeric Nano Formulations. Int. J. Biol. Macromol. 2020, 164, 2197–2203.
- Makvandi, P.; Baghbantaraghdari, Z.; Zhou, W.; Zhang, Y.; Manchanda, R.; Agarwal, T.; Wu, A.; Maiti, T.K.; Varma, R.S.; Smith, B.R. Gum polysaccharide/nanometal hybrid biocomposites in cancer diagnosis and therapy. Biotechnol. Adv. 2021, 48, 107711.
- Hajipour, A.R.; Khorsandi, Z.; Mortazavi, M.; Farrokhpour, H. Green, efficient and large-scale synthesis of benzimidazoles, benzoxazoles and benzothiazoles derivatives using ligand-free cobalt-nanoparticles: As potential anti-estrogen breast cancer agents, and study of their interactions with estrogen receptor by molecular docking. RSC Adv. 2015, 5, 107822–107828.
- Khorsandi, Z.; Hajipour, A.R.; Sarfjoo, M.R.; Varma, R.S. A Pd/Cu-Free magnetic cobalt catalyst for C–N cross coupling reactions: Synthesis of abemaciclib and fedratinib. Green Chem. 2021, 23, 5222–5229.
- Khorsandi, Z.; Keshavarzipour, F.; Varma, R.S.; Hajipour, A.R.; Sadeghi-Aliabadi, H. Sustainable synthesis of potential antitumor new derivatives of Abemaciclib and Fedratinib via C-N cross coupling reactions using Pd/Cu-free Co-catalyst. Mol. Catal. 2021, 517, 112011.
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer. 2005, 5, 761–772.
- Jego, G.; Hazoumé, A.; Seigneuric, R.; Garrido, C. Targeting heat shock proteins in cancer. Cancer Lett. 2013, 332, 275–285.
- Calderwood, S.K.; Khaleque, M.A.; Sawyer, D.B.; Ciocca, D.R. Heat Shock Proteins in Cancer: Chaperones of Tum Origenesis. J. Pet. Sci. Eng. 2006, 31, 164–172.
- Patel, H.J.; Patel, P.D.; Ochiana, S.O.; Yan, P.; Sun, W.; Patel, M.R.; Shah, S.K.; Tramentozzi, E.; Brooks, J.; Bolaender, A.J. Structure–Activity Relationship in a Purine-Scaffold Compound Series with Selectivity for the Endoplasmic Reticulum Hsp90 ParalogGrp94. Med. Chem. 2015, 58, 3922–3943.
- Johnson, J.L. Evolution and function of diverse Hsp90 homologs and cochaperone proteins. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 607–613.
- Csermely, P.; Miyata, Y.; Schnaider, T.; Yahara, I. Autophosphorylation of grp94 (Endoplasmin). J. Biol. Chem. 1995, 270, 6381–6388.
- Mishra, P.; Bolon, D.N. Designed Hsp90 Heterodimers Reveal an Asymmetric ATPase-Driven Mechanism In Vivo. Mol. Cell 2014, 53, 344–350.
- Meyer, P.; Prodromou, C.; Hu, B.; Vaughan, C.; Roe, S.M.; Panaretou, B.; Piper, P.W.; Pearl, L.H. Structural and Functional Analysis of the Middle Segment of Hsp90: Implications for ATP Hydrolysis and Client Protein and Cochaperone Interactions. Mol. Cell 2003, 11, 647–658.
- Gupta, S.D. Hsp90 Flexibility and Development of its Inhibitors for the Treatment of Cancer. Curr. Chem. Biol. 2018, 12, 53–64.
- Gupta, S.D. Novel Anti-Cancer Drugs Based on Hsp90 Inhibitory Mechanisms: A Recent Report. In Medicinal Chemistry with Pharma-Ceutical Product Development; Apple Academic Press: New York, NY, USA, 2019; pp. 57–104.
- Gupta, S.D.; Pan, C.H. Recent update on discovery and development of Hsp90 inhibitors as senolytic agents. Int. J. Biol. Macromol. 2020, 161, 1086–1098.
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Vartholomaiou, E.; Echeverría, P.C.; Picard, D. Unusual Suspects in the Twilight Zone Between the Hsp90 Interactome and Carcinogenesis. Adv. Cancer Res. 2016, 129, 1–30.
- Hayat, U.; Elliott, G.T.; Olszanski, A.J.; Altieri, D.C. Feasibility and safety of targeting mitochondria for cancer therapy–preclinical characterization of gamitrinib, a first-in-class, mitochondriaL-targeted small molecule Hsp90 inhibitor. Cancer Biol. Ther. 2022, 23, 117–126.
- Biondini, M.; Kiepas, A.; El-Houjeiri, L.; Annis, M.G.; Hsu, B.E.; Fortier, A.-M.; Morin, G.; Martina, J.A.; Sirois, I.; Aguilar-Mahecha, A.; et al. HSP90 inhibitors induce GPNMB cell-surface expression by modulating lysosomal positioning and sensitize breast cancer cells to glembatumumab vedotin. Oncogene 2022, 41, 1701–1717.
- Epp-Ducharme, B.; Dunne, M.; Fan, L.; Evans, J.C.; Ahmed, L.; Bannigan, P.; Allen, C. Heat-activated nanomedicine formulation improves the anticancer potential of the HSP90 inhibitor luminespib in vitro. Sci. Rep. 2021, 11, 11103.
- Wu, Y.-W.; Chao, M.-W.; Tu, H.-J.; Chen, L.-C.; Hsu, K.-C.; Liou, J.-P.; Yang, C.-R.; Yen, S.-C.; HuangFu, W.-C.; Pan, S.-L. A novel dual HDAC and HSP90 inhibitor, MPT0G449, downregulates oncogenic pathways in human acute leukemia in vitro and in vivo. Oncogenesis 2021, 10, 39.
- Mshaik, R.; Simonet, J.; Georgievski, A.; Jamal, L.; Bechoua, S.; Ballerini, P.; Bellaye, P.-S.; Mlamla, Z.; de Barros, J.-P.P.; Geissler, A.; et al. HSP90 inhibitor NVP-BEP800 affects stability of SRC kinases and growth of T-cell and B-cell acute lymphoblastic leukemias. Blood Cancer J. 2021, 11, 61.
- Abdelmoaty, A.A.A.; Zhang, P.; Lin, W.; Fan, Y.-J.; Ye, S.-N.; Xu, J.-H. C0818, a novel curcumin derivative, induces ROS-dependent cytotoxicity in human hepatocellular carcinoma cells in vitro via disruption of Hsp90 function. Acta Pharmacol. Sin. 2022, 43, 446–456.
- Zhao, S.; Zhou, L.; Dicker, D.T.; Lev, A.; Zhang, S.; Ross, E.; El-Deiry, W.S. Anti-Cancer Efficacy Including Rb-Deficient Tumors and VHL-Independent HIF1α Proteasomal Destabilization by Dual Targeting of CDK1 or CDK4/6 and Hsp90. Sci. Rep. 2021, 11, 20871.
- Chen, X.-L.; Liu, P.; Zhu, W.-L.; Lou, L.-G. DCZ5248, a novel dual inhibitor of Hsp90 and autophagy, exerts antitumor activity against colon cancer. Acta Pharmacol. Sin. 2021, 42, 132–141.
- Cheng, C.-J.; Liu, K.-X.; Zhang, M.; Shen, F.-K.; Ye, L.-L.; Wu, W.-B.; Hou, X.-T.; Hao, E.-W.; Hou, Y.-Y.; Bai, G. Okicamelliaside targets the N-terminal chaperone pocket of HSP90 disrupts the chaperone protein interaction of HSP90-CDC37 and exerts antitumor activity. Acta Pharmacol. Sin. 2021, 43, 1046–1058.
- Konstantinopoulos, P.A.; Cheng, S.-C.; Supko, J.G.; Polak, M.; Wahner-Hendrickson, A.E.; Ivy, S.P.; Bowes, B.; Sawyer, H.; Basada, P.; Hayes, M.; et al. Combined PARP and HSP90 inhibition: Preclinical and Phase 1 evaluation in patients with advanced solid tumours. Br. J. Cancer 2021, 126, 1027–1036.
- Magwenyane, A.M.; Lawal, M.M.; Amoako, D.G.; Somboro, A.M.; Agoni, C.; Khan, R.B.; Mhlongo, N.N.; Kumalo, H.M. Exploring the inhibitory mechanism of resorcinylic isoxazole amine NVP-AUY922 towards the discovery of potential heat shock protein 90 (Hsp90) inhibitors. Sci. Afr. 2022, 15, e01107.
- Serwetnyk, M.A.; Blagg, S.J. The Disruption of Protein−Protein Interactions with Co-chaperones and Client Substrates as a Strategy Towards Hsp90 Inhibition. Acta. Pharm. Sin. B 2021, 11, 1446–1468.
- Roe, S.M.; Prodromou, C.; O’Brien, R.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. Structural Basis for Inhibition of the Hsp90 Molecular Chaperone by the Antitumor Antibiotics Radicicol and Geldanamycin. J. Med. Chem. 1999, 42, 260–266.
- Supko, J.G.; Hickman, R.L.; Grever, M.R.; Malspeis, L. Preclinical Pharmacologic Evaluation of Geldanamycin as an Antitumor Agent. Cancer Chemother. Pharmacol. 1995, 36, 305–315.
- Banerji, U.; O’Donnell, A.; Scurr, M.; Benson, C.; Hanwell, J.; Clark, S.; Raynaud, F.; Turner, A.; Walton, M.; Workman, P.; et al. Phase I Trial of the Heat Shock Protein 90 (Hsp90) Inhibitor 17-Allylamino 17-Demethoxygeldanamycin (17AAG). Pharmacokinetic (PK) Profile and Pharmacodynamic (PD) Endpoints. Proc. Am. Soc. Clin. Oncol. 2001, 20, 326.
- Egorin, M.J.; Zuhowski, E.G.; Rosen, D.M.; Sentz, D.L.; Covey, J.M.; Eiseman, J.L. Plasma pharmacokinetics and tissue distribution of 17-(allylamino)-17-demethoxygeldanamycin (NSC 330507) in CD2F1 mice1. Cancer Chemother. Pharmacol. 2001, 47, 291–302.
- Egorin, M.J.; Lagattuta, T.F.; Hamburger, D.R.; Covey, J.M.; White, K.D.; Musser, S.M.; Eiseman, J.L. Pharmacokinetics, tissue distribution, and metabolism of 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin (NSC 707545) in CD2F1 mice and Fischer 344 rats. Cancer Chemother. Pharmacol. 2002, 49, 7–19.
- Munster, P.N.; Tong, L.; Schwartz, L.; Larson, S.; Kenneson, K.; De La Cruz, A.; Rosen, N.; Scher, H. Phase I Trial of 17(Allylamino)-17-demethoxygeldanamycin (17AAG) in Patients (Pts) with Advanced Solid Malignancies. Proc. Am. Soc. Clin. Oncol. 2001, 20, 83–91.
- Soga, S.; Sharma, S.V.; Shiotsu, Y.; Shimizu, M.; Tahara, H.; Yamaguchi, K.; Ikuina, Y.; Murakata, C.; Tamaoki, T.; Kurebayashi, J.; et al. Stereospecific antitumor activity of radicicol oxime derivatives. Cancer Chemother. Pharmacol. 2001, 48, 435–445.
- Johnson, V.A.; Singh, E.K.; Nazarova, L.A.; Alexander, L.D.; McAlpine, S.R. Macrocyclic inhibitors of hsp90. Curr. Top. Med. Chem. 2010, 10, 1380–1402.
- Porter, J.R.; Ge, J.; Lee, J.; Normant, E.; West, K. Ansamycin Inhibitors of Hsp90: Nature’s Prototype for Anti-chaperone Therapy. Curr. Top. Med. 2009, 9, 1386–1418.
- Tanida, S.; Hasegawa, T.; Higashide, E.; Macbecins, I.; Macbecins, I.I. New Antitumor Antibiotics. I. Producing Organism, Fermentation and Antimicrobial Activities. J. Antibiot. 1980, 33, 199–204.
- Tian, Z.-Q.; Liu, Y.; Zhang, D.; Wang, Z.; Dong, S.D.; Carreras, C.W.; Zhou, Y.; Rastelli, G.; Santi, D.V.; Myles, D.C. Synthesis and biological activities of novel 17-aminogeldanamycin derivatives. Bioorg. Med. Chem. 2004, 12, 5317–5329.
- Wagner, A.J.; Chugh, R.; Rosen, L.S.; Morgan, J.A.; George, S.; Gordon, M.; Dunbar, J.; Normant, E.; Grayzel, D.; Demetri, G.D. A Phase I Study of the HSP90 Inhibitor Retaspimycin Hydrochloride (IPI-504) in Patients with Gastrointestinal Stromal Tumors or Soft-Tissue Sarcomas. Clin. Cancer Res. 2013, 19, 6020–6029.
- Floris, G.; Debiec-Rychter, M.; Wozniak, A.; Stefan, C.; Normant, E.; Faa, G.; Machiels, K.; Vanleeuw, U.; Sciot, R.; Schöffski, P. The Heat Shock Protein 90 Inhibitor IPI-504 Induces KIT Degradation, Tumor Shrinkage, and Cell Proliferation Arrest in Xenograft Models of Gastrointestinal Stromal Tumors. Mol. Cancer Ther. 2011, 10, 1897–1908.
- Floris, G.; Sciot, R.; Wozniak, A.; Van Looy, T.; Wellens, J.; Faa, G.; Normant, E.; Debiec-Rychter, M.; Schöffski, P. The Novel Hsp90 inhibitor, IPI-493, is Highly Effective in Human Gastrostrointestinal Stromal Tumor Xenografts Carrying Heterogeneous KIT Mutations. Clin. Cancer Res. 2011, 17, 5604–5614.
- Whitesell, L.; Mimnaugh, E.G.; De Costa, B.; Myers, C.E.; Neckers, L.M. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: Essential role for stress proteins in oncogenic transformation. Proc. Natl. Acad. Sci. USA 1994, 91, 8324–8328.
- Stebbins, C.E.; Russo, A.A.; Schneider, C.; Rosen, N.; Hartl, F.; Pavletich, N.P. Crystal Structure of an Hsp90–Geldanamycin Complex: Targeting of a Protein Chaperone by an Antitumor Agent. Cell 1997, 89, 239–250.
- Soga, S.; Shiotsu, Y.; Akinaga, S.; Sharma, S. Development of Radicicol Analogues. Curr. Cancer Drug Targets 2003, 3, 359–369.
- Miyata, Y. Hsp90 Inhibitor Geldanamycin and Its Derivatives as Novel Cancer Chemotherapeutic Agents. Curr. Pharm. Des. 2005, 11, 1131–1138.
- Chiosis, G.; Lucas, B.; Huezo, H.; Solit, D.; Basso, A.; Rosen, N. Development of Purine-Scaffold Small Molecule Inhibitors of Hsp90. Curr. Cancer Drug Targets 2003, 3, 371–376.
- Conejo-García, A.; García-Rubiño, M.E.; Marchal, J.A.; Núñez, M.C.; Ramírez, A.; Cimino, S.; García, M.; Aránega, A.; Gallo, M.A.; Campos, J.M. Synthesis and anticancer activity of (RS)-9-(2,3-dihydro-1,4-benzoxaheteroin-2-ylmethyl)-9H-purines. Eur. J. Med. Chem. 2011, 46, 3795–3801.
- Pelliccia, S.; Amato, J.; Capasso, D.; Di Gaetano, S.; Massarotti, A.; Piccolo, M.; Irace, C.; Tron, G.C.; Pagano, B.; Randazzo, A.J. Bio-Inspired Dual-Selective BCL-2/c-MYC G-Quadruplex Binders: Design, Synthesis, and Anticancer Activity of Drug-like Imidazo purine Derivatives. Med. Chem. 2019, 63, 2035–2050.
- Ashour, F.A.; Rida, S.M.; El-Hawash, S.A.; ElSemary, M.M.; Badr, M.H. Synthesis, Anticancer, Anti-HIV-1, and Antimicrobial Activity of Some Tricyclic Triazino and Triazolo purine Derivatives. Cancer Causes Control 2012, 21, 1107–1119.
- Kinali-Demirci, S.; İdil, Ö.; Dişli, A. Synthesis of Some Novel Purine Derivatives Incorporating Tetrazole Ring and Investigation of their Antimicrobial Activity and DNA Interactions. Cancer Cell Int. J. Mol. Histol. 2015, 24, 1218–1225.
- Abdallah, A.E.; Elgemeie, G.H. Development; Therapy, Design, Synthesis, Docking, and Antimicrobial Evaluation of Some Novel Pyrazolo Pyrimidines and their Corresponding Cycloalkane Ring-fused Derivatives as Purine Analogs. Drug Des. Devel. Ther. 2018, 12, 1785–1798.
- Rosemeyer, H. The Chemodiversity of Purine as a Constituent of Natural Products. Chem. Biodivers. 2004, 1, 361–401.
- Kaneko, K.; Aoyagi, Y.; Fukuuchi, T.; Inazawa, K.; Yamaoka, N. Total Purine and Purine Base Content of Common Foodstuffs for Facilitating Nutritional Therapy for Gout and Hyperuricemia. Biol. Pharm. Bull. 2014, 37, 709–721.
- Yin, J.; Ren, W.; Huang, X.; Deng, J.; Li, T.; Yin, Y. Potential Mechanisms Connecting Purine Metabolism and Cancer Therapy. Front. Immunol. 2018, 9, 1697.
- Chiosis, G.; Timaul, M.N.; Lucas, B.; Munster, P.N.; Zheng, F.F.; Sepp-Lorenzino, L.; Rosen, N. A small molecule designed to bind to the adenine nucleotide pocket of Hsp90 causes Her2 degradation and the growth arrest and differentiation of breast cancer cells. Chem. Biol. 2001, 8, 289–299.
- Bao, R.; Lai, C.-J.; Qu, H.; Wang, D.; Yin, L.; Zifcak, B.; Atoyan, R.; Wang, J.; Samson, M.; Forrester, J.; et al. CUDC-305, a Novel Synthetic HSP90 Inhibitor with Unique Pharmacologic Properties for Cancer Therapy. Clin. Cancer Res. 2009, 15, 4046–4057.
- Zhang, H.; Neely, L.; Lundgren, K.; Yang, Y.-C.; Lough, R.; Timple, N.; Burrows, F. BIIB021, a synthetic HSP90 inhibitor, has broad application against tumors with acquired multidrug resistance. Int. J. Cancer 2010, 126, 1226–1234.
- Yin, X.; Zhang, H.; Lundgren, K.; Wilson, L.; Burrows, F.; Shores, C.G. BIIB021, a novel HSP90 inhibitor, sensitizes head and neck squamous cell carcinoma to radiotherapy. Int. J. Cancer 2010, 126, 1216–1225.
- Chaurasiya, A.; Wahan, S.K.; Sahu, C.; Chawla, P.A. An insight into the rational design of recent purine-based scaffolds in targeting various cancer pathways. J. Mol. Struct. 2022, 134308.
- Wang, X.-T.; Bao, C.-H.; Jia, Y.-B.; Wang, N.; Ma, W.; Liu, F.; Wang, C.; Wang, J.-B.; Song, Q.-X.; Cheng, Y.-F. BIIB021, a novel Hsp90 inhibitor, sensitizes esophageal squamous cell carcinoma to radiation. Biochem. Biophys. Res. Commun. 2014, 452, 945–950.
- ElFiky, A.; Saif, M.W.; Beeram, M.; Brien, S.O.; Lammanna, N.; Castro, J.E.; Woodworth, J.; Perea, R.; Storgard, C.; Von Hoff, D.D. BIIB021, an oral, synthetic non-ansamycin Hsp90 inhibitor: Phase I experience. J. Clin. Oncol. 2008, 26, 2503.
- Dickson, M.A.; Okuno, S.H.; Keohan, M.L.; Maki, R.G.; D’Adamo, D.R.; Akhurst, T.J.; Antonescu, C.R.; Schwartz, G.K. Phase II study of the HSP90-inhibitor BIIB021 in gastrointestinal stromal tumors. Ann. Oncol. 2013, 24, 252–257.
- Emami, S.; Dadashpour, S. Current developments of coumarin-based anti-cancer agents in medicinal chemistry. Eur. J. Med. Chem. 2015, 102, 611–630.
- Zhang, L.; Xu, Z. Coumarin-containing hybrids and their anticancer activities. Eur. J. Med. Chem. 2019, 181, 111587.
- Al-Warhi, T.; Sabt, A.; Elkaeed, E.B.; Eldehna, W.M. Recent advancements of coumarin-based anticancer agents: An up-to-date review. Bioorg. Chem. 2020, 103, 104163.
- Song, X.; Fan, J.; Liu, L.; Liu, X.; Gao, F. Coumarin derivatives with anticancer activities: An update. Arch. Pharm. 2020, 353, 2000025.
- Qin, H.-L.; Zhang, Z.-W.; Ravindar, L.; Rakesh, K. Antibacterial activities with the structure-activity relationship of coumarin derivatives. Eur. J. Med. Chem. 2020, 207, 112832.
- Keshavarzipour, F.; Tavakol, H. The synthesis of coumarin derivatives using choline chloride/zinc chloride as a deep eutectic solvent. J. Iran. Chem. Soc. 2016, 13, 149–153.
- Ahmed, E.Y.; Elserwy, W.S.; El-Mansy, M.F.; Serry, A.M.; Salem, A.M.; Abdou, A.M.; Abdelrahman, B.A.; Elsayed, K.H.; Elaziz, M.R.A. Angiokinase inhibition of VEGFR-2, PDGFR and FGFR and cell growth inhibition in lung cancer: Design, synthesis, biological evaluation and molecular docking of novel azaheterocyclic coumarin derivatives. Bioorg. Med. Chem. Lett. 2021, 48, 128258.
- Burlison, J.A.; Neckers, L.; Smith, A.B.; Maxwell, A.A.; Blagg, B.S.J. Novobiocin: Redesigning a DNA Gyrase Inhibitor for Selective Inhibition of Hsp90. J. Am. Chem. Soc. 2006, 128, 15529–15536.
- Shelton, S.N.; Shawgo, M.E.; Matthews, S.B.; Lu, Y.; Donnelly, A.C.; Szabla, K.; Tanol, M.; Vielhauer, G.A.; Rajewski, R.A.; Matts, R.L.; et al. KU135, a Novel Novobiocin-Derived C-Terminal Inhibitor of the 90-kDa Heat Shock Protein, Exerts Potent Antiproliferative Effects in Human Leukemic Cells. Mol. Pharmacol. 2009, 76, 1314–1322.
- Garg, G.; Zhao, H.; Blagg, B.S.J. Design, Synthesis, and Biological Evaluation of Ring-Constrained Novobiocin Analogues as Hsp90 C-Terminal Inhibitors. ACS Med. Chem. Lett. 2015, 6, 204–209.
- Wei, Q.; Ning, J.-Y.; Dai, X.; Gao, Y.-D.; Su, L.; Zhao, B.-X.; Miao, J.-Y. Discovery of novel HSP90 inhibitors that induced apoptosis and impaired autophagic flux in A549 lung cancer cells. Eur. J. Med. Chem. 2018, 145, 551–558.
- Kouznetsov, V.V.; Méndez, L.Y.V.; Galvis, C.E.P.; Villamizar, M.C.O. The direct C–H alkenylation of quinoline N-oxides as a suitable strategy for the synthesis of promising antiparasitic drugs. New J. Chem. 2020, 44, 12–19.
- Keshavarzipour, F.; Tavakol, H. Zinc Cation Supported on Carrageenan Magnetic Nanoparticles: A Novel, Green and Efficient Catalytic System for One-pot Three-Component Synthesis of Quinoline Derivatives. Appl. Organomet. Chem. 2017, 31, 3682–3692.
- Insuasty, D.; García, S.; Abonia, R.; Insuasty, B.; Quiroga, J.; Nogueras, M.; Laali, K.K. Design, Synthesis, and Molecular Docking Study of Novel Quinoline-Based Bis-Chalcones as Potential Antitumor Agents. Arch. Pharm. 2021, 354, 2100094–2100103.
- Jin, G.; Li, Z.; Xiao, F.; Qi, X.; Sun, X. Optimization of activity localization of quinoline derivatives: Design, synthesis, and dual evaluation of biological activity for potential antitumor and antibacterial agents. Bioorg. Chem. 2020, 99, 103837.
- Mishra, S.; Salahuddin, R.K.; Majumder, A.; Kumar, A.; Singh, C.; Tiglani, D. Updates on Synthesis and Biological Activities of Quinoline Derivatives: A Review. Int. J. Pharm. Sci. Rev. Res. 2021, 13, 3941–3960.
- Goyal, S.; Binnington, B.; McCarthy, S.D.; Desmaële, D.; Férrié, L.; Figadère, B.; Loiseau, P.M.; Branch, D.R. Inhibition of in vitro Ebola infection by anti-parasitic quinoline derivatives. F1000Research 2020, 9, 268.
- Verma, C.; Quraishi, M.; Ebenso, E.E. Quinoline and its derivatives as corrosion inhibitors: A review. Surfaces Interfaces 2020, 21, 100634.
- Kayamba, F.; Malimabe, T.; Ademola, I.K.; Pooe, O.J.; Kushwaha, N.D.; Mahlalela, M.; van Zyl, R.L.; Gordon, M.; Mudau, P.T.; Zininga, T.; et al. Design and synthesis of quinoline-pyrimidine inspired hybrids as potential plasmodial inhibitors. Eur. J. Med. Chem. 2021, 217, 113330.
- Rao, K.V.; Cullen, W.P. Streptonigrin, an Antitumor Substance. I. Isolation and Characterization. Antibiot. Annu. 1959, 7, 950–953.
- Chirigos, M.A.; Pearson, J.W.; Papas, T.S.; Woods, W.A.; Wood, H.B., Jr.; Spahn, G. Effect of Three Strains of BeG Against a Murine Leukemia After Drug Therapy. Cancer Chemother. Rep. 1973, 57, 305–309.
- Balitz, D.M.; Bush, J.A.; Bradner, W.T.; Doyle, T.W.; O’Herron, F.A.; Nettleton, D.E. Isolation of lavendamycin. A new antibiotic from Streptomyces lavendulae. J. Antibiot. 1982, 35, 259–265.
- Ganesh, T.; Min, J.; Thepchatri, P.; Du, Y.; Li, L.; Lewis, I.; Wilson, L.; Fu, H.; Chiosis, G.; Dingledine, R.; et al. Discovery of aminoquinolines as a new class of potent inhibitors of heat shock protein 90 (Hsp90): Synthesis, biology, and molecular modeling. Bioorg. Med. Chem. 2008, 16, 6903–6910.
- Audisio, D.; Messaoudi, S.; Cegielkowski, L.; Peyrat, J.-F.; Brion, J.-D.; Methy-Gonnot, D.; Radanyi, C.; Renoir, J.-M.; Alami, M. Discovery and Biological Activity of 6BrCaQ as an Inhibitor of the Hsp90 Protein Folding Machinery. Chem. Med. Chem. 2011, 6, 804–815.
- Nepali, K.; Kumar, S.; Huang, H.-L.; Kuo, F.-C.; Lee, C.-H.; Kuo, C.-C.; Yeh, T.-K.; Li, Y.-H.; Chang, J.-Y.; Liou, J.-P.; et al. 2-Aroylquinoline-5,8-diones as potent anticancer agents displaying tubulin and heat shock protein 90 (HSP90) inhibition. Org. Biomol. Chem. 2015, 14, 716–723.
- Malayeri, S.O.; Abnous, K.; Arab, A.; Akaberi, M.; Mehri, S.; Zarghi, A.; Ghodsi, R. Design, Synthesis and Biological Evaluation of 7-(Aryl)-2,3-dihydro- dioxino Quinoline Derivatives as Potential Hsp90 Inhibitors and Anticancer Agents. Bioorg. Med. Chem. 2017, 25, 1294–1302.
- Liang, C.; Hao, H.; Wu, X.; Li, Z.; Zhu, J.; Lu, C.; Shen, Y. Design and synthesis of N-(5-chloro-2,4-dihydroxybenzoyl)-(R)-1,2,3,4-tetrahydroisoquinoline-3-carboxamides as novel Hsp90 inhibitors. Eur. J. Med. Chem. 2016, 121, 272–282.
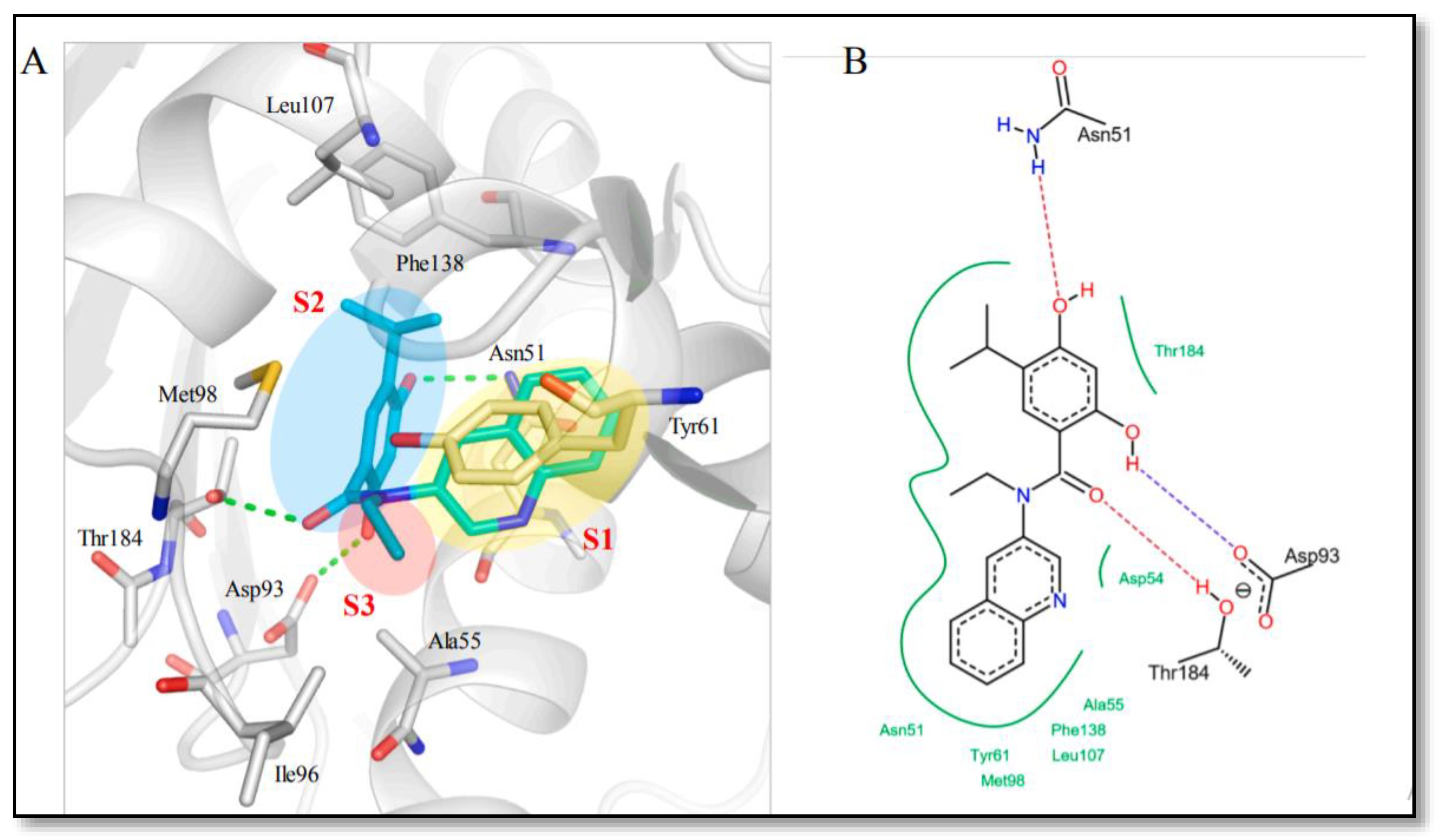
- Nepali, K.; Lin, M.-H.; Chao, M.-W.; Peng, S.-J.; Hsu, K.-C.; Lin, T.E.; Chen, M.-C.; Lai, M.-J.; Pan, S.-L.; Liou, J.-P. Amide-tethered quinoline-resorcinol conjugates as a new class of HSP90 inhibitors suppressing the growth of prostate cancer cells. Bioorg. Chem. 2019, 91, 103119.
- Relitti, N.; Saraswati, A.P.; Chemi, G.; Brindisi, M.; Brogi, S.; Herp, D.; Schmidtkunz, K.; Saccoccia, F.; Ruberti, G.; Ulivieri, C.; et al. Novel quinolone-based potent and selective HDAC6 inhibitors: Synthesis, molecular modeling studies and biological investigation. Eur. J. Med. Chem. 2021, 212, 112998.


