 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mohd Heikal Mohd Yunus | + 2385 word(s) | 2385 | 2020-11-24 07:19:31 | | | |
| 2 | Vivi Li | -79 word(s) | 2306 | 2020-12-02 06:54:44 | | |
Video Upload Options
Osteoarthritis (OA) is the most well-known disease among the geriatric and the main cause of significant disability in daily living. It has a multifactorial aetiology and characterized by pathological changes of knee joint structure including cartilage erosion, synovial inflammation and subchondral sclerosis with osteophyte formation. To date, no efficient treatment is capable of altering the pathological progression of OA. This topic described significant mediators such as cytokines, proteolytic enzymes, and nitric oxide, that trigger the loss of the normal homeostasis and structural changes in the articular cartilage during the progression of OA.
1. Introduction
Osteoarthritis (OA) is the most common cause of chronic joint pain among the geriatric population. OA is defined as the progressive deterioration of articular cartilage, followed by inflammation in the synovial cavity. Due to the extreme pain in the joint caused by OA, patients experience significant disability in their daily living.
Prior to the 1990s, OA had been described as cartilage wear and tear, where the articular cartilage is degraded due to incremental pressure on a particular joint. With the advancement of molecular biology, the paradigm of OA pathophysiology has shifted to it being described as an inflammatory joint disease [1][2][3]. This follows the discovery of several inflammatory mediators that actuate chondrocytes to produce matrix metalloproteinases (MMPs), a major player in articular matrix degradation.
In more recent years, the establishment of a direct correlation between age-related inflammation and the disturbance in gut microbiota has brought attention to the gut–joint axis hypothesis of OA. The link between the disturbance in gut microbiota, defined as gut dysbiosis, and OA has been demonstrated in several studies [4].
Maintenance of the articular cartilage is tightly regulated by the anabolic and catabolic pathways of the cartilage matrix. In a healthy joint, the articular chondrocytes adapt to the various stresses to which they are subjected by altering their metabolism, resulting in the degradation or synthesis of the cartilage matrix to suit the demands of the body [5][6][7].
The complex pathogenesis of OA comprises the interplay of numerous factors ranging from hereditary inclination to alteration of gene expression via changes in the mechanical loading experienced by articular chondrocytes [5]. Dysregulation in these molecular repertoires can prompt the deterioration of the articular cartilage and the risk of progression into OA, either directly or indirectly [1].
2. Pathological Changes in OA
Considering its complexity, the initiation, progression, and severity of OA are each driven by a plethora of factors. Furthermore, in all individuals, OA does not progress at a similar rate. At the cartilage–bone interface, an inverse relationship between subchondral bone changes and articular cartilage degeneration has been reported. As the subchondral bone thickens, a higher stage of cartilage degeneration is observed [8].
The earliest pathological changes in OA are commonly seen on the articular cartilage surface, with fibrillation occurring in focal regions experiencing maximal load. The proliferation of chondrocytes, the only cell type present in cartilage, dramatically accelerates in response to the loss of matrix. Some chondrocytes undergo a phenotypic change to hypertrophic chondrocytes, which is similar to the cells found in the growth plate’s hypertrophic zones. As OA progresses, extensive matrix degradation and loss occurs due to the continuous production of proteases driven by proinflammatory cytokines, which stimulate chondrocytes to produce more cytokines and proteases in an autocrine and paracrine manner. As significant matrix damage occurs, areas of the matrix devoid of cells can be seen as a result of chondrocyte apoptosis.
The bone changes in OA include subchondral sclerosis due to increased collagen production, with osteophyte formation and bone cysts at more advanced stages. Osteophytes have been described as bone and cartilage outgrowths occurring at the joint area. The direction of osteophyte growth is sensitive to the size and local cartilage narrowing, except for the lateral tibia and medial patella [9]. Biomechanical factors support osteophyte development. Most patients with symptomatic OA exhibit synovial inflammation and hypertrophy [10]. However, synovitis inflammation is not the triggering factor for primary OA, but contributes to the progression of pain and disease [11].
Plain radiographs underestimate the joint tissue involvement in OA, since they only visualize a component of the condition including cartilage loss that result in joint space narrowing and bony changes that result in subchondral sclerosis, cysts, and osteophyte formation. Once these changes are apparent on radiographs, the condition has significantly advanced [12].
Magnetic resonance imaging (MRI) studies can detect early disease and have provided evidence of matrix changes in cartilage, synovitis, bone marrow lesions, and degenerative changes in soft-tissue structures beyond the cartilage including ligaments and the knee menisci [13].
The arthroscope can play an important diagnostic role in patients with unexplained knee pain and swelling or in patients with established knee arthritis whose symptoms are disproportionate to radiographic findings [14].
Moreover, apart from these above mentioned pathological changes, the paradigm has shifted to the involvement of various inflammatory mediators, proteinases, cell proliferation, and biochemical parameters in the development of the disease.
3. Inflammatory Mediators
3.1. Cytokines and Chemokines
Inflammatory mediators such as cytokines are the key component of most inflammatory processes. Accordingly, a multitude of cytokines have been associated with OA pathogenesis. In OA patients, cartilage matrix homeostasis is disrupted by proinflammatory cytokines and chemokines [15][16]. Investigation of the cytokines and chemokines involved during OA progression revealed the upregulation of IL-1, IL-6, and IL-8 [17][18][19].
These cytokines act as both autocrine and paracrine agents, to stimulate the collective production of proteases, nitric oxide (NO), and eicosanoids such as prostaglandins and leukotrienes by macrophages and chondrocytes. Subsequently, the action of these inflammatory mediators in the cartilage results in the induction of the catabolic pathways, inhibition of matrix synthesis, and promotion of cellular apoptosis [19]. The cellular apoptosis, particularly in chondrocytes, is driven by the inhibition of autophagy by the proinflammatory cytokines [20][21].
The production of IL-1 by the stimulated chondrocytes in turn induces the synthesis of MMPs, namely MMP-1, MMP-3 and MMP-13. This is accompanied by the amplification of proinflammatory cytokines such as TNF-α, IL-6 and the chemokine IL-8, which magnifies the cartilage matrix breakdown effects in the catabolic cascade, further enhancing articular chondrocyte destruction [17][22]. IL-1 has also been proposed to contribute to the decline in cartilage matrix by inhibiting the synthesis of key components of ECM, such as proteoglycans, aggrecan, and type II collagen [23][24][25].
Moreover, the involvement of fibronectin in cartilage degradation is also apparent when fragments of the protein induce the expression of inflammatory cytokines, chemokines, and MMPs in chondrocytes [26][27]. In normal adult cartilage, chondrocytes synthesize matrix components very slowly. Finally, chondrocyte senescence is the other major contributor to OA development and progression. This is due to the senescent cells’ loss of the capacity for maintaining and repairing the cartilage ECM [28]. Both IL-6 and IL-8 are a key cytokine and chemokine, respectively, also known to be secreted by senescent cells, which is known as the senescence-associated secretory phenotype [29].
3.2. Proteases
The MMP family plays a major role in articular cartilage homeostasis. Collagenases (MMP-1, MMP-13) are responsible for degradation of the collagenous framework, whereas stromelysin (MMP-3) and aggrecanase (ADAMT-4), which is responsible for proteoglycan degradation, play prominent roles in ECM degradation [16][30]. The inflammatory cytokines synthesized by OA chondrocytes, i.e., IL-1 and TNF-α, can trigger increased MMP expression, suppress MMP enzyme inhibitors, and decrease ECM synthesis. Active stromelysin serves as an activator of collagenase 1, 2, and 3 (MMP-1, MMP-8, and MMP-13, respectively) implicated in type II collagen degradation [23][24][25].
MMP-13, the protease that preferentially degrades type II collagen, may be the most important in OA progression, considering type II collagen as the main collagen type in ECM. Indeed, MMP-13 expression is of greatly increased in OA [19][31]. In contrast to MMP-1 and MMP-3, which are present in high levels in OA synovial fluid, MMP-13 is highly expressed in OA cartilage, indicating its important role in the degradation of human articular cartilage throughout OA [30][31][32]. Moreover, only hypertrophic chondrocytes express the MMP-13 encoding genes, which can all be detected in OA cartilage [27].
Taken together, cytokine regulation of the equilibrium between the anabolic and catabolic processes determines the integrity of articular joint tissue. In pathogenesis, the occurrence of anabolic activity overwhelms that of catabolic activity, resulting in tissue degeneration [33].
3.3. Inflammatory Mediator Enzymes
Other than cytokines and proteases, the expression of enzymes such as inducible NO synthase (iNOS), which generates the free radical NO, and cyclooxygenase-2 (COX- 2), which produces prostaglandin E2 (PGE2), are also altered in OA [22]. Here, the proinflammatory cytokine IL-1 stimulates the upregulation of both PGE2 and NO by inducing the gene expression or activity of COX-2 and iNOS [23].
Akhtar et al. (2011) noted that IL-1, together with mechanical loading of the cartilage, induced upregulation of the iNOS gene, which in turn increased the NO production. NO contributes to articular degradation by upregulating synthesis of MMP via cyclic GMP (cGMP)-dependent pathways while simultaneously inhibiting the synthesis of both proteoglycans and collagen [34][35].
Notably, NO has also been implicated to play a role in mediating chondrocyte apoptosis, a common feature in progressive OA [19][33][36]. Moreover, NO also alters mitochondrial function in OA chondrocytes, resulting in reduced cell survival by inhibiting the activity of the mitochondrial respiratory chain and ATP synthesis [37].
COX activation enhances the production of MMP-3 while inhibiting proteoglycans and collagen synthesis and inducing chondrocyte apoptosis [37]. With IL-1 stimulation, the chondrocyte is upregulated, eventually leading to increased production of PGE2 [38]. Martel-Pelletier et al. (2003) suggested the role of PGE2 in inflammation, apoptosis, angiogenesis, and probably the structural changes characterizing arthritic diseases [39]. Increased PGE2 production causes cartilage resorption by suppressing proteoglycans production, enhancing the degradation of both aggrecan and type II collagen and potentiating the effects of other inflammatory mediators such as IL-6 and MMP-13 [40].
Even without cytokine stimulation, when cultured in vitro, both NO and COX-2 levels are already highly expressed in chondrocytes from OA tissues [41][42]. Such metabolic changes may indicate a permanent phenotypical shift in OA chondrocytes. Moreover, the discovery of COX-2-induced PGE2 in fibrocartilage implies a role for PGE2 in the secondary remodeling of the tissue that causes osteophyte formation of in the pathogenesis of OA [43].
Matriptase, a novel protease found in OA articular chondrocytes, initiates cartilage matrix degradation by activating proteinase-activated receptor 2 (PAR-2) [44]. The absence of PAR-2 results in the absence of OA-associated pain and osteophyte formation [45]. Therefore, it is possible that the PAR-2 system is involved in the inflammatory response-mediated ECM degradation in OA. In addition, secreted proinflammatory cytokines up-regulate the expression of PAR-2, inducing greater production of proinflammatory cytokines (IL-6, IL-8), metalloproteinases, and PGE2 to enhance the inflammatory responses [46][47]. Boileau et al. (2007) demonstrated that PAR-2 expression and protein levels in OA chondrocytes have increased significantly and that the levels are regulated by the proinflammatory cytokine IL-1. The PAR-2 activation resulting in increased rates of MMPs (MMP-1, MMP-13) and COX-2 indicates that it could play a key role in the catabolic and inflammatory pathways during the progression of OA by inducing major catabolic and inflammatory mediators [47][48][49][50]. The interaction of various inflammatory mediators in the event of OA is summarized in Table 2 and Figure 1.
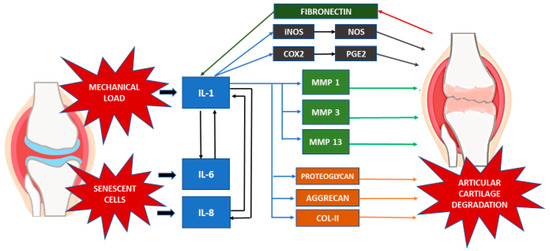
Figure 1. Inflammatory mediators in OA.
Table 2. Potential mechanisms in the event of OA.
| Inflammatory Mediators | Description of Mechanism |
|---|---|
| Cytokines and Chemokines | IL-1, IL-6, IL-8:
|
| Proteases | MMP-1, -3, -13 and ADAMT-4:
|
| iNOS (NO) |
|
| COX-2 (PGE2) |
|
| PAR-2 |
|
3.4. Other Potential Mediators of OA
Several studies have provided evidence for a number of potential mediators that induce OA but are not considered as inflammatory mediators. These factors also induce activating pathways that promote joint tissue destruction or inhibiting the ability of cells to repair damaged matrix of OA.
Hypoxia-inducible factor 1-alpha (HIF-1α) is an important mediator of cellular response towards an oxygen-deprived environment. Articular cartilage resides in an environment that is devoid of oxygen. Homeostasis of this tissue is mainly maintained by the HIF-1α regulatory mechanism. Single nucleotide polymorphism (SNP) studies have revealed that a defect in HIF-1α disrupts the catabolic pathways of the cartilage matrix. Instead of undergoing autophagy, defects of HIF-1α resulted in chondrocyte hypertrophy in response to the hypoxia environment [55].
The Wnt signaling pathways play a substantial role in the joint development. In OA, the interaction between the underlying subchondral bone and articular cartilage brought about the hypothesis of Wnt signaling pathways role in OA [56]. In a mouse model, activation of the Wnt signaling pathway in subchondral bone induces degradation of the articular cartilage. This reiterates the potential role of the Wnt signaling pathway in the pathogenesis of OA [57].
Nerve growth factor (NGF) is a neurotrophin that transmits the pain information following inflammation. In bovine chondrocytes induced with TGF-β1 and IL-1β, NGF expression was found to be elevated. The elevated expression of NGF is mediated by activin receptor-like kinase 5 (ALK5) and the Smad 2/3 complex [58]. When cartilage explant was incubated with osteoarthritic synovium, TGF-β1 and Smad 2/3 were inhibited, suggesting a potential inhibition of NGF [59]. In conclusion, NGF might be an important mediator to the OA event.
References
- Nguyen, L.T.; Sharma, A.R.; Chakraborty, C.; Saibaba, B.; Ahn, M.E.; Lee, S.S. Review of Prospects of Biological Fluid Biomarkers in Osteoarthritis. Int. J. Mol. Sci. 2017, 18, 601.
- Berenbaum, F. Osteoarthritis as an inflammatory disease (osteoarthritis is not osteoarthrosis). Osteoarthr. Cartil. 2013, 21, 16–21.
- Jeong, J.-H.; Moon, S.-J.; Jhun, J.-Y.; Yang, E.-J.; Cho, M.-L.; Min, J.-K. Eupatilin Exerts Antinociceptive and Chondroprotective Properties in a Rat Model of Osteoarthritis by Downregulating Oxidative Damage and Catabolic Activity in Chondrocytes. PLoS ONE 2015, 10, e0130882.
- De Sire, A.; De Sire, R.; Petito, V.; Masi, L.; Cisari, C.; Gasbarrini, A.; Scaldaferri, F.; Invernizzi, M. Gut–Joint Axis: The Role of Physical Exercise on Gut Microbiota Modulation in Older People with Osteoarthritis. Nutrients 2020, 12, 574.
- Akhtar, N.; Miller, M.J.S.; Haqqi, T.M. Effect of a Herbal-Leucine mix on the IL-1b induced cartilage degradation and inflammatory gene expression in human chondrocytes. BMC Complement. Altern. Med. 2011, 11, 66.
- Jeffrey, J.E.; Aspden, R.M. Cyclooxygenase inhibition lowers prostaglandin E2 release from articular cartilage and reduces apoptosis but not proteoglycan degradation following an impact load in vitro. Arthritis Res. Ther. 2007, 9, R129.
- Falah, M.; Nierenberg, G.; Soudry, M.; Hayden, M.; Volpin, G. Treatment of articular cartilage lesions of the knee. Int. Orthop. 2010, 34, 621–630.
- Bobinac, D.; Spanjol, J.; Zoricic, S.; Maric, I. Changes in articular cartilage and subchondral bone histomorphometry in osteoarthritic knee joints in humans. Bone 2003, 32, 284–290.
- Nagaosa, Y.; Lanyon, P.; Doherty, M. Characterisation of size and direction of osteophyte in knee osteoarthritis: A radiographic study. Ann. Rheum. Dis. 2002, 61, 319–324.
- Baker, K.; Grainger, A.; Niu, J.; Clancy, M.; Guermazi, A.; Crema, M.; Hughes, L.; Buckwalter, J.; Wooley, A.; Nevitt, M.; et al. Relation of synovitis to knee pain using contrast-enhanced MRIs. Ann. Rheum. Dis. 2010, 69, 1779–1783.
- Wang, X.; Hunter, D.J.; Jin, X.; Ding, C. The importance of synovial inflammation in osteoarthritis: Current evidence from imaging assessments and clinical trials. Osteoarthr. Cartil. 2018, 26, 165–174.
- Loeser, R.F.; Goldring, S.R.; Scanzello, C.R.; Goldring, M.B. Osteoarthritis: A disease of the joint as an organ. Arthritis Rheum. 2012, 64, 1697–1707.
- Sharma, L.; Chmiel, J.S.; Almagor, O.; Dunlop, D.; Guermazi, A.; Bathon, J.M.; Eaton, C.B.; Hochberg, M.C.; Jackson, R.D.; Kwoh, C.K.; et al. Significance of Preradiographic Magnetic Resonance Imaging Lesions in Persons at Increased Risk of Knee Osteoarthritis. Arthritis Rheumatol. 2014, 66, 1811–1819.
- O’Rourke, K.S.; Ike, R.W. Diagnostic arthroscopy in the arthritis patient. Rheum. Dis. Clin. N. Am. 1994, 20, 321–342.
- Chow, Y.Y.; Chin, K.-Y. The Role of Inflammation in the Pathogenesis of Osteoarthritis. Mediat. Inflamm. 2020, 2020, 1–19.
- Akkiraju, H.; Nohe, A. Role of Chondrocytes in Cartilage Formation, Progression of Osteoarthritis and Cartilage Regeneration. J. Dev. Biol. 2015, 3, 177–192.
- Hoff, P.; Buttgereit, F.; Burmester, G.-R.; Jakstadt, M.; Gaber, T.; Andreas, K.; Matziolis, G.; Perka, C.; Röhner, E. Osteoarthritis synovial fluid activates pro-inflammatory cytokines in primary human chondrocytes. Int. Orthop. 2012, 37, 145–151.
- Kapoor, M.; Martel-Pelletier, J.; Lajeunesse, D.; Pelletier, J.P.; Fahmi, H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 33–42.
- Abramson, S.B.; Straub, R.H. Developments in the scientific understanding of osteoarthritis. Arthritis Res. Ther. 2009, 11, 227.
- Hwang, H.S.; Kim, H.A. Chondrocyte Apoptosis in the Pathogenesis of Osteoarthritis. Int. J. Mol. Sci. 2015, 16, 26035–26054.
- Caramés, B.; Hasegawa, A.; Taniguchi, N.; Miyaki, S.; Blanco, F.J.; Lotz, M. Autophagy activation by rapamycin reduces severity of experimental osteoarthritis. Ann. Rheum. Dis. 2012, 71, 575–581.
- Wojdasiewicz, P.; Poniatowski, L.A.; Szukiewicz, D. The Role of Inflammatory and Anti-Inflammatory Cytokines in the Pathogenesis of Osteoarthritis. Mediat. Inflamm. 2014, 2014, 561459.
- Goldring, M.B.; Otero, M. Inflammation in osteoarthritis. Curr. Opin. Rheumatol. 2014, 23, 471–478.
- Ruszymah, B.H.I.; Shamsul, B.; Chowdhury, S.R.; Hamdan, M. Effect of cell density on formation of three-dimensional cartilaginous constructs using fibrin & human osteoarthritic chondrocytes. Indian J. Med. Res. 2019, 149, 641–649.
- Ude, C.C.; Sulaiman, S.B.; Min-Hwei, N.; Chen, H.C.; Ahmad, J.; Yahaya, N.M.; Saim, A.B.; Idrus, R.B.H. Cartilage Regeneration by Chondrogenic Induced Adult Stem Cells in Osteoarthritic Sheep Model. PLoS ONE 2014, 9, e98770.
- Fichter, M.; Körner, U.; Schomburg, J.; Jennings, L.; Cole, A.A.; Mollenhauer, J. Collagen degradation products modulate matrix metalloproteinase expression in cultured articular chondrocytes. J. Orthop. Res. 2005, 24, 63–70.
- Houard, X.; Goldring, M.B. Berenbaum Francis, Homeostatic Mechanisms in Articular Cartilage and Role of Inflammation in Osteoarthritis. Curr. Rheumatol. Rep. 2013, 15, 375.
- Musumeci, G.; Aiello, F.C.; Szychlinska, M.A.; Di Rosa, M.; Castrogiovanni, P.; Mobasheri, A. Osteoarthritis in the XXIst Century: Risk Factors and Behaviours that Influence Disease Onset and Progression. Int. J. Mol. Sci. 2015, 16, 6093–6112.
- Tsuchida, A.I.; Beekhuizen, M.; Rutgers, M.; van Osch, G.J.; Bekkers, J.E.; Bot, A.G.; Geurts, B.; Dhert, W.J.; Saris, D.B.; Creemers, L.B. Cytokine profiles in the joint depend on pathology, but are different between synovial fluid, cartilage tissue and cultured chondrocytes. Arthritis Res. Ther. 2014, 16, 441.
- Martel-Pelletier, J. Pathophysiology of osteoarthritis. Osteoarthr. Cartil. 2004, 12, S31–S33.
- Blasioli, D.J.; Kaplan, D.L. The Roles of Catabolic Factors in the Development of Osteoarthritis. Tissue Eng. Part B 2014, 20, 355–363.
- Yin, J.; Yang, Z.; Cao, Y.-P.; Ge, Z. Characterization of human primary chondrocytes of osteoarthritic cartilage at varying severity. Chin. Med. J. 2011, 124, 4245–4253.
- Sandell, L.; Aigner, T. Articular cartilage and changes in arthritis. An introduction: Cell biology of osteoarthritis. Arthritis Res. 2001, 3, 107–113.
- Abramson, S.B. Osteoarthritis and nitric oxide. Osteoarthr. Cartil. 2008, 16, S15–S20.
- Ridnour, L.A.; Windhausen, A.N.; Isenberg, J.S.; Yeung, N.; Thomas, D.D.; Vitek, M.P.; Roberts, D.D.; Wink, D.A. Nitric oxide regulates matrix metalloproteinase-9 activity by guanylylcyclase-dependent and -independent pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 16898–16903.
- Sun, L.; Wang, X.; Kaplan, D.L. A 3D cartilage—Inflammatory cell culture system for the modeling of human osteoarthritis. Biomaterials 2011, 32, 5581–5589.
- Lee, A.S.; Ellman, M.B.; Yan, D.; Kroin, J.S.; Cole, B.J.; Van Wijnen, A.J.; Im, H.-J. A current review of molecular mechanisms regarding osteoarthritis and pain. Gene 2013, 527, 440–447.
- Shimpo, H.; Sakai, T.; Kondo, S.; Mishima, S.; Yoda, M.; Hiraiwa, H.; Ishiguro, N. Regulation of prostaglandin E2 synthesis in cells derived from chondrocytes of patients with osteoarthritis. J. Orthop. Sci. 2009, 14, 611–617.
- Martel-Pelletier, J.; Pelletier, J.-P.; Fahmi, H. Cyclooxygenase-2 and prostaglandins in articular tissues. Semin. Arthritis Rheum. 2003, 33, 155–167.
- Wang, P.; Zhu, F.; Konstantopoulos, K. Interleukin-6 Synthesis in Human Chondrocytes Is Regulated via the Antagonistic Actions of Prostaglandin (PG)E2 and 15-deoxy-Δ12,14-PGJ2. PLoS ONE 2011, 6, e27630.
- Yang, K.G.A.; Saris, D.B.; Geuze, R.E.; Van Rijen, M.H.P.; Van Der Helm, Y.J.M.; Verbout, A.J.; Creemers, L.B.; Dhert, W.J. Altered in vitro chondrogenic properties of chondrocytes harvested from unaffected cartilage in osteoarthritic joints. Osteoarthr. Cartil. 2006, 14, 561–570.
- Maldonado, M.; Nam, J. The Role of Changes in Extracellular Matrix of Cartilage in the Presence of Inflammation on the Pathology of Osteoarthritis. BioMed. Res. Int. 2013, 2013, 1–10.
- Hardy, M.M.; Seibert, K.; Manning, P.T.; Currie, M.G.; Woerner, B.M.; Edwards, D.; Koki, A.; Tripp, C.S. Cyclooxygenase 2-dependent prostaglandin E2 modulates cartilage proteoglycan degradation in human osteoarthritis explants. Arthritis Rheum. 2002, 46, 1789–1803.
- Milner, J.M.; Patel, A.; Davidson, R.K.; Swingler, T.E.; Désilets, A.; Young, D.A.; Kelso, E.B.; Donell, S.T.; Cawston, T.E.; Clark, I.M.; et al. Matriptase is a novel initiator of cartilage matrix degradation in osteoarthritis. Arthritis Rheum. 2010, 62, 1955–1966.
- Huesa, C.; Ortiz, A.C.; Dunning, L.; McGavin, L.; Bennett, L.; McIntosh, K.; Crilly, A.; Kurowska-Stolarska, M.; Plevin, R.; van’t Hof, R.J.; et al. Proteinase-activated receptor 2 modulates OA-related pain, cartilage and bone pathology. Ann. Rheum. Dis. 2015, 75, 1989–1997.
- Xiang, Y.; Masuko-Hongo, K.; Sekine, T.; Nakamura, H.; Yudoh, K.; Nishioka, K.; Kato, T. Expression of proteinase-activated receptors (PAR)-2 in articular chondrocytes is modulated by IL-1beta, TNF-alpha and TGF-beta. Osteoarthr. Cartil. 2006, 14, 1163–1173.
- Chen, T.L.; Lin, Y.F.; Cheng, C.W.; Chen, S.Y.; Sheu, M.T.; Leung, T.K.; Qin, C.H.; Chen, C.H. Anti-Inflammatory mechanisms of the proteinase activated receptor 2-inhibiting peptide in human synovial cells. J. Biomed. Sci. 2011, 18, 43. [PubMed]
- Boileau, C.; Amiable, N.; Martel-Pelletier, J.; Fahmi, H.; Duval, N.; Pelletier, J.-P. Activation of proteinase-activated receptor 2 in human osteoarthritic cartilage upregulates catabolic and proinflammatory pathways capable of inducing cartilage degradation: A basic science study. Arthritis Res. Ther. 2007, 9, R121.
- McIntosh, K.A.; Plevin, R.; Ferrell, W.R.; Lockhart, J.C. The therapeutic potential of proteinase-activated receptors in arthritis. Curr. Opin. Pharmacol. 2007, 7, 334–338.
- MY, M.H.; Ahmad Nazrun, S.; Busra, M.F.; Chua, K.H.; Norzana, A.G.; Rizal, A.R. Pro-Chondrogenic Propensity of Sticopus Chloronotus Aqueous Extracts on Human Osteoarthritis Articular Chondrocytes In Vitro. Indian J. Med. Res. Pharm. Sci. 2017, 4, 37–50.
- Chia, Y.C.; Beh, H.C.; Ng, C.J.; Teng, C.L.; Hanafi, N.S.; Choo, W.Y.; Ching, S.M. Ethnic differences in the prevalence of knee pain among adults of a community in a cross-sectional study. BMJ Open 2016, 6, e011925.
- Biver, E.; Berenbaum, F.; Valdes, A.M.; De Carvalho, I.A.; Bindels, L.B.; Brandi, M.L.; Calder, P.C.; Castronovo, V.; Cavalier, E.; Cherubini, A.; et al. Gut microbiota and osteoarthritis management: An expert consensus of the European society for clinical and economic aspects of osteoporosis, osteoarthritis and musculoskeletal diseases (ESCEO). Ageing Res. Rev. 2019, 55, 100946.
- Ezengin, A.; Eprentice, A.; Ward, K.A. Ethnic Differences in Bone Health. Front. Endocrinol. 2015, 6, 24.
- Allen, K. Racial and ethnic disparities in osteoarthritis phenotypes. Curr. Opin. Rheumatol. 2010, 22, 528–532.
- Pfander, D.; Gelse, K. Hypoxia and osteoarthritis: How chondrocytes survive hypoxic environments. Curr. Opin. Rheumatol. 2007, 19, 457–462.
- Luyten, F.P.; Tylzanowski, P.; Lories, R.J. Wnt signaling and osteoarthritis. Bone 2009, 44, 522–527.
- Davidson, E.B.; Vitters, E.L.; Bennink, M.B.; van Lent, P.L.E.M.; van Caam, A.P.M.; Blom, A.B.; van den Berg, W.B.; van de Loo, F.A.J.; van der Kraan, P.M. Inducible chondrocyte-specific overexpression of BMP2 in young mice results in severe aggravation of osteophyte formation in experimental OA without altering cartilage damage. Ann. Rheum. Dis. 2015, 74, 1257–1264.
- Matsunobu, T.; Torigoe, K.; Ishikawa, M.; De Vega, S.; Kulkarni, A.B.; Iwamoto, Y.; Yamada, Y. Critical Roles of the TGF-β type I Receptor ALK5 in Perichondrial Formation and Function, Cartilage Integrity, and Osteoblast Differentiation during Growth Plate Development. Dev. Biol. 2009, 332, 325–338.
- Madej, W.; Buma, P.; van der Kraan, P. Inflammatory Conditions Partly Impair the Mechanically Mediated Activation of Smad2/3 Signaling in Articular Cartilage. Arth. Res. Ther. 2016, 18, 146.

