 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Beatrix Zheng | -- | 2262 | 2022-11-03 01:31:35 |
Video Upload Options
Ras is a family of related proteins which is expressed in all animal cell lineages and organs. All Ras protein family members belong to a class of protein called small GTPase, and are involved in transmitting signals within cells (cellular signal transduction). Ras is the prototypical member of the Ras superfamily of proteins, which are all related in 3D structure and regulate diverse cell behaviours. When Ras is 'switched on' by incoming signals, it subsequently switches on other proteins, which ultimately turn on genes involved in cell growth, differentiation and survival. Mutations in ras genes can lead to the production of permanently activated Ras proteins. As a result, this can cause unintended and overactive signaling inside the cell, even in the absence of incoming signals. Because these signals result in cell growth and division, overactive Ras signaling can ultimately lead to cancer. The 3 Ras genes in humans (HRas, KRas, and NRas) are the most common oncogenes in human cancer; mutations that permanently activate Ras are found in 20% to 25% of all human tumors and up to 90% in certain types of cancer (e.g., pancreatic cancer). For this reason, Ras inhibitors are being studied as a treatment for cancer and other diseases with Ras overexpression.
1. History
The first two ras genes, HRAS and KRAS, were identified[1] from studies of two cancer-causing viruses, the Harvey sarcoma virus and Kirsten sarcoma virus, by Edward M. Scolnick and colleagues at the National Institutes of Health (NIH).[2] These viruses were discovered originally in rats during the 1960s by Jennifer Harvey[3] and Werner Kirsten,[4] respectively, hence the name Rat sarcoma.[1] In 1982, activated and transforming human ras genes were discovered in human cancer cells by Geoffrey M. Cooper at Harvard,[5] Mariano Barbacid and Stuart A. Aaronson at the NIH,[6] Robert Weinberg at MIT,[7] and Michael Wigler at Cold Spring Harbor Laboratory.[8] A third ras gene was subsequently discovered by researchers in the group of Robin Weiss at the Institute of Cancer Research,[9][10] and Michael Wigler at Cold Spring Harbor Laboratory,[11] named NRAS, for its initial identification in human neuroblastoma cells.
The three human ras genes encode extremely similar proteins made up of chains of 188 to 189 amino acids. Their gene symbols are HRAS, NRAS and KRAS, the latter of which produces the K-Ras4A and K-Ras4B isoforms from alternative splicing.
2. Structure

Ras contains six beta strands and five alpha helices.[12] It consists of two domains, a G domain of 166 amino acids, about 20kDa, that binds guanosine nucleotides, and a C-terminal membrane targeting region (CAAX-COOH, also known as CAAX box), which is lipid-modified by farnesyl transferase, RCE1 and ICMT.
The G domain contains five G motifs that bind GDP/GTP directly. The G1 motif, or the P-loop, binds the beta phosphate of GDP and GTP. The G2 motif, also called Switch I, contains threonine35, which binds the terminal phosphate (γ-phosphate) of GTP and the divalent magnesium ion bound in the active site. The G3 motif, also called Switch II, has a DXXGQ motif. The D is aspartate57, which is specific for guanine versus adenine binding, and Q is glutamine61, the crucial residue that activates a catalytic water molecule for hydrolysis of GTP to GDP. The G4 motif contains a LVGNKxDL motif, and provides specific interaction to guanine. The G5 motif contains a SAK consensus sequence. The A is alanine146, which provides specificity for guanine rather than adenine.
The two switch motifs, G2 and G3, are the main parts of the protein that move upon activation by GTP. This conformational change by the two switch motifs is what mediates the basic functionality as a molecular switch protein. This GTP-bound state of Ras is the "on" state, and the GDP-bound state is the "off" state.
Ras also binds a magnesium ion which helps to coordinate nucleotide binding.
3. Function
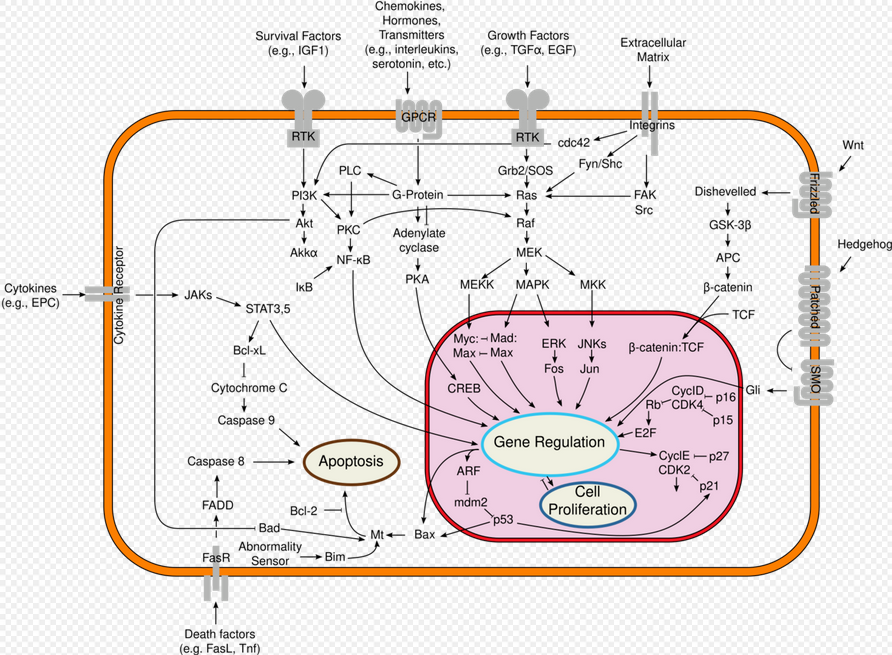
Ras proteins function as binary molecular switches that control intracellular signaling networks. Ras-regulated signal pathways control such processes as actin cytoskeletal integrity, cell proliferation, cell differentiation, cell adhesion, apoptosis, and cell migration. Ras and Ras-related proteins are often deregulated in cancers, leading to increased invasion and metastasis, and decreased apoptosis.
Ras activates several pathways, of which the mitogen-activated protein (MAP) kinase cascade has been well-studied. This cascade transmits signals downstream and results in the transcription of genes involved in cell growth and division.[13] Another Ras-activated signaling pathway is the PI3K/AKT/mTOR pathway, which stimulates protein synthesis and cellular growth, and inhibits apoptosis.
3.1. Activation and Deactivation
Ras is a G protein, or a guanosine-nucleotide-binding protein. Specifically, it is a single-subunit small GTPase, which is related in structure to the Gα subunit of heterotrimeric G proteins (large GTPases). G proteins function as binary signaling switches with "on" and "off" states. In the "off" state it is bound to the nucleotide guanosine diphosphate (GDP), while in the "on" state, Ras is bound to guanosine triphosphate (GTP), which has an extra phosphate group as compared to GDP. This extra phosphate holds the two switch regions in a "loaded-spring" configuration (specifically the Thr-35 and Gly-60). When released, the switch regions relax which causes a conformational change into the inactive state. Hence, activation and deactivation of Ras and other small G proteins are controlled by cycling between the active GTP-bound and inactive GDP-bound forms.
The process of exchanging the bound nucleotide is facilitated by guanine nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs). As per its classification, Ras has an intrinsic GTPase activity, which means that the protein on its own will hydrolyze a bound GTP molecule into GDP. However this process is too slow for efficient function, and hence the GAP for Ras, RasGAP, may bind to and stabilize the catalytic machinery of Ras, supplying additional catalytic residues ("arginine finger") such that a water molecule is optimally positioned for nucleophilic attack on the gamma-phosphate of GTP. An inorganic phosphate is released and the Ras molecule is now bound to a GDP. Since the GDP-bound form is "off" or "inactive" for signaling, GTPase Activating Protein inactivates Ras by activating its GTPase activity. Thus, GAPs accelerate Ras inactivation.
GEFs catalyze a "push and pull" reaction which releases GDP from Ras. They insert close to the P-loop and magnesium cation binding site and inhibit the interaction of these with the gamma phosphate anion. Acidic (negative) residues in switch II "pull" a lysine in the P-loop away from the GDP which "pushes" switch I away from the guanine. The contacts holding GDP in place are broken and it is released into the cytoplasm. Because intracellular GTP is abundant relative to GDP (approximately 10 fold more)[13] GTP predominantly re-enters the nucleotide binding pocket of Ras and reloads the spring. Thus GEFs facilitate Ras activation.[12] Well known GEFs include Son of Sevenless (Sos) and cdc25 which include the RasGEF domain.
The balance between GEF and GAP activity determines the guanine nucleotide status of Ras, thereby regulating Ras activity.
In the GTP-bound conformation, Ras has a high affinity for numerous effectors which allow it to carry out its functions. These include PI3K. Other small GTPases may bind adaptors such as arfaptin or second messenger systems such as adenylyl cyclase. The Ras binding domain is found in many effectors and invariably binds to one of the switch regions, because these change conformation between the active and inactive forms. However, they may also bind to the rest of the protein surface.
Other proteins exist may change the activity of Ras family proteins. One example is GDI (GDP Disassociation Inhibitor); These function by slowing the exchange of GDP for GTP and thus, prolonging the inactive state of Ras family members. Other proteins that augment this cycle may exist.
3.2. Membrane Attachment
Ras is attached to the cell membrane owing to its prenylation and palmitoylation (HRAS and NRAS) or the combination of prenylation and a polybasic sequence adjacent to the prenylation site (KRAS). The C-terminal CaaX box of Ras first gets farnesylated at its Cys residue in the cytosol, allowing Ras to loosely insert into the membrane of the endoplasmatic reticulum and other cellular membranes. The Tripeptide (aaX) is then cleaved from the C-terminus by a specific prenyl-protein specific endoprotease and the new C-terminus is methylated by a methyltransferase. KRas processing is completed at this stage. Dynamic electrostatic interactions between its positively charged basic sequence with negative charges at the inner leaflet of the plasma membrane account for its predominant localization at the cell surface at steady-state. NRAS and HRAS are further processed on the surface of the Golgi apparatus by palmitoylation of one or two Cys residues, respectively, adjacent to the CaaX box. The proteins thereby become stably membrane anchored (lipid-rafts) and are transported to the plasma membrane on vesicles of the secretory pathway. Depalmitoylation by acyl-protein thioesterases eventually releases the proteins from the membrane, allowing them to enter another cycle of palmitoylation and depalmitoylation.[14] This cycle is believed to prevent the leakage of NRAS and HRAS to other membranes over time and to maintain their steady-state localization along the Golgi apparatus, secretory pathway, plasma membrane and inter-linked endocytosis pathway.
4. Members
The clinically most notable members of the Ras subfamily are HRAS, KRAS and NRAS, mainly for being implicated in many types of cancer.[15]
However, there are many other members of this subfamily as well:[16] DIRAS1; DIRAS2; DIRAS3; ERAS; GEM; MRAS; NKIRAS1; NKIRAS2; NRAS; RALA; RALB; RAP1A; RAP1B; RAP2A; RAP2B; RAP2C; RASD1; RASD2; RASL10A; RASL10B; RASL11A; RASL11B; RASL12; REM1; REM2; RERG; RERGL; RRAD; RRAS; RRAS2
5. Ras in Cancer
Mutations in the Ras family of proto-oncogenes (comprising H-Ras, N-Ras and K-Ras) are very common, being found in 20% to 30% of all human tumors.[15] it is reasonable to speculate that a pharmacological approach that curtails Ras activity may represent a possible method to inhibit certain cancer types. Ras point mutations are the single most common abnormality of human proto-oncogenes.[17] Ras inhibitor trans-farnesylthiosalicylic acid (FTS, Salirasib) exhibits profound anti-oncogenic effects in many cancer cell lines.[18][19]
5.1. Inappropriate Activation
Inappropriate activation of the gene has been shown to play a key role in improper signal transduction, proliferation and malignant transformation.[13]
Mutations in a number of different genes as well as RAS itself can have this effect. Oncogenes such as p210BCR-ABL or the growth receptor erbB are upstream of Ras, so if they are constitutively activated their signals will transduce through Ras.
The tumour suppressor gene NF1 encodes a Ras-GAP – its mutation in neurofibromatosis will mean that Ras is less likely to be inactivated. Ras can also be amplified, although this only occurs occasionally in tumours.
Finally, Ras oncogenes can be activated by point mutations so that the GTPase reaction can no longer be stimulated by GAP – this increases the half life of active Ras-GTP mutants.[20]
5.2. Constitutively Active Ras
Constitutively active Ras (RasD) is one which contains mutations that prevent GTP hydrolysis, thus locking Ras in a permanently 'On' state.
The most common mutations are found at residue G12 in the P-loop and the catalytic residue Q61.
- The glycine to valine mutation at residue 12 renders the GTPase domain of Ras insensitive to inactivation by GAP and thus stuck in the "on state". Ras requires a GAP for inactivation as it is a relatively poor catalyst on its own, as opposed to other G-domain-containing proteins such as the alpha subunit of heterotrimeric G proteins.
- Residue 61[21] is responsible for stabilizing the transition state for GTP hydrolysis. Because enzyme catalysis in general is achieved by lowering the energy barrier between substrate and product, mutation of Q61 to K necessarily reduces the rate of intrinsic Ras GTP hydrolysis to physiologically meaningless levels.
See also "dominant negative" mutants such as S17N and D119N.
5.3. Ras-Targeted Cancer Treatments
Reovirus was noted to be a potential cancer therapeutic when studies suggested it reproduces well in certain cancer cell lines. It replicates specifically in cells that have an activated Ras pathway (a cellular signaling pathway that is involved in cell growth and differentiation).[22] Reovirus replicates in and eventually kills Ras-activated tumour cells and as cell death occurs, progeny virus particles are free to infect surrounding cancer cells. This cycle of infection, replication and cell death is believed to be repeated until all tumour cells carrying an activated Ras pathway are destroyed.
Another tumor-lysing virus that specifically targets tumor cells with an activated Ras pathway is a type II herpes simplex virus (HSV-2) based agent, designated FusOn-H2.[23] Activating mutations of the Ras protein and upstream elements of the Ras protein may play a role in more than two-thirds of all human cancers, including most metastatic disease. Reolysin, a formulation of reovirus, and FusOn-H2 are currently in clinical trials or under development for the treatment of various cancers.[24] In addition, a treatment based on siRNA anti-mutated K-RAS (G12D) called siG12D LODER is currently in clinical trials for the treatment of locally advanced pancreatic cancer (NCT01188785, NCT01676259).[25]
In glioblastoma mouse models SHP2 levels were heightened in cancerous brain cells. Inhibiting SHP2 in turn inhibited Ras dephosphorylation. This reduced tumor sizes and accompanying rise in survival rates.[26][27]
Other strategies have attempted to manipulate the regulation of the above-mentioned localization of Ras. Farnesyltransferase inhibitors have been developed to stop the farnesylation of Ras and therefore weaken its affinity to membranes.[28] Other inhibitors are targeting the palmitoylation cycle of Ras through inhibiting depalmitoylation by acyl-protein thioesterases, potentially leading to a destabilization of the Ras cycle.[29]
References
- "RAS oncogenes: the first 30 years". Nat. Rev. Cancer 3 (6): 459–65. June 2003. doi:10.1038/nrc1097. PMID 12778136. https://dx.doi.org/10.1038%2Fnrc1097
- "Human genome contains four genes homologous to transforming genes of Harvey and Kirsten murine sarcoma viruses". Proc. Natl. Acad. Sci. U.S.A. 79 (16): 4848–52. August 1982. doi:10.1073/pnas.79.16.4848. PMID 6289320. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=346782
- Harvey JJ (December 1964). "An unidentified virus which causes the rapid production of tumours in mice". Nature 204 (4963): 1104–5. doi:10.1038/2041104b0. PMID 14243400. https://dx.doi.org/10.1038%2F2041104b0
- "Properties of a murine sarcoma virus". Bibl Haematol (36): 246–9. 1970. PMID 5538357. http://www.ncbi.nlm.nih.gov/pubmed/5538357
- Cooper GM (August 1982). "Cellular transforming genes". Science 217 (4562): 801–6. doi:10.1126/science.6285471. PMID 6285471. https://dx.doi.org/10.1126%2Fscience.6285471
- "T24 human bladder carcinoma oncogene is an activated form of the normal human homologue of BALB- and Harvey-MSV transforming genes". Nature 298 (5872): 343–7. July 1982. doi:10.1038/298343a0. PMID 6283384. https://dx.doi.org/10.1038%2F298343a0
- "Human EJ bladder carcinoma oncogene is homologue of Harvey sarcoma virus ras gene". Nature 297 (5866): 474–8. June 1982. doi:10.1038/297474a0. PMID 6283357. https://dx.doi.org/10.1038%2F297474a0
- "Activation of the T24 bladder carcinoma transforming gene is linked to a single amino acid change". Nature 300 (5894): 762–5. December 1982. doi:10.1038/300762a0. PMID 7177195. https://dx.doi.org/10.1038%2F300762a0
- "A transforming gene present in human sarcoma cell lines". Nature 299 (5879): 171–3. September 1982. doi:10.1038/299171a0. PMID 6287287. https://dx.doi.org/10.1038%2F299171a0
- "Identification of transforming gene in two human sarcoma cell lines as a new member of the ras gene family located on chromosome 1". Nature 303 (5916): 396–400. 1983. doi:10.1038/303396a0. PMID 6304521. https://dx.doi.org/10.1038%2F303396a0
- "Isolation and preliminary characterization of the transforming gene of a human neuroblastoma cell line". PNAS 80 (2): 383–7. January 1983. doi:10.1073/pnas.80.2.383. PMID 6300838. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=393381
- "The guanine nucleotide-binding switch in three dimensions". Science 294 (5545): 1299–304. November 2001. doi:10.1126/science.1062023. PMID 11701921. https://dx.doi.org/10.1126%2Fscience.1062023
- "Chapter 25, Cancer". Molecular cell biology (4th ed.). San Francisco: W.H. Freeman. 2000. ISBN 0-7167-3706-X.
- "Spatio-temporal segregation of Ras signals: one ship, three anchors, many harbors". Current Opinion in Cell Biology 18 (4): 351–7. 2006. doi:10.1016/j.ceb.2006.06.007. PMID 16781855. https://dx.doi.org/10.1016%2Fj.ceb.2006.06.007
- Bos J (1989). "ras oncogenes in human cancer: a review". Cancer Res 49 (17): 4682–9. PMID 2547513. http://www.ncbi.nlm.nih.gov/pubmed/2547513
- "The Ras superfamily at a glance". J. Cell Sci. 118 (Pt 5): 843–6. March 2005. doi:10.1242/jcs.01660. PMID 15731001. https://dx.doi.org/10.1242%2Fjcs.01660
- Robbins and Cotran (2010). Pathologic Basis of Disease 8th ed.. p. 282.
- "The Ras inhibitor farnesylthiosalicylic acid (Salirasib) disrupts the spatiotemporal localization of active Ras: a potential treatment for cancer.". Methods Enzymol 439: 467–89. 2008. doi:10.1016/S0076-6879(07)00432-6. PMID 18374183. https://dx.doi.org/10.1016%2FS0076-6879%2807%2900432-6
- Roy Blum; yoel kloog (2005). "Ras Inhibition in Glioblastoma Down-regulates Hypoxia-Inducible Factor-1, Causing Glycolysis Shutdown and Cell Death". Cancer Research 65 (3): 999–1006. PMID 15705901. http://www.ncbi.nlm.nih.gov/pubmed/15705901
- "Targeting the Ras signaling pathway: a rational, mechanism-based treatment for hematologic malignancies?". Blood 96 (5): 1655–69. 2000. PMID 10961860. http://www.ncbi.nlm.nih.gov/pubmed/10961860
- Omim - Neuroblastoma Ras Viral Oncogene Homolog; Nras https://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=164790&a=164790_AllelicVariant0002
- "Reovirus: Rationale and clinical trial update". Curr. Opin. Mol. Ther. 11 (5): 532–9. October 2009. PMID 19806501. http://www.ncbi.nlm.nih.gov/pubmed/19806501
- Fu, Xinping; Prigge-J, Cai-R; Xiaoliu Zhang. "A mutant type 2 herpes simplex virus deleted for the protein kinase domain of the ICP10 gene is a potent oncolytic virus". Molecular Therapy 13 (5): 882–890. doi:10.1016/j.ymthe.2006.08.180. https://dx.doi.org/10.1016%2Fj.ymthe.2006.08.180
- "Oncolytic viral therapy using reovirus". Methods Mol. Biol. 542: 607–34. 2009. doi:10.1007/978-1-59745-561-9_31. PMID 19565924. https://dx.doi.org/10.1007%2F978-1-59745-561-9_31
- "ClinicalTrials.gov". http://www.clinicaltrials.gov/ct2/results?term=silenseed.
- Bunda, Severa; Burrell, Kelly; Heir, Pardeep; Zeng, Lifan; Alamsahebpour, Amir; Kano, Yoshihito; Raught, Brian; Zhang, Zhong-Yin et al. (2015-11-30). "Inhibition of SHP2-mediated dephosphorylation of Ras suppresses oncogenesis" (in en). Nature Communications 6: 8859. doi:10.1038/ncomms9859. PMID 26617336. PMC 4674766. http://www.nature.com/ncomms/2015/151130/ncomms9859/full/ncomms9859.html.
- Taub, Ben (2015-12-03). "Scientists Find Way To Deactivate Most Common Cancer-Causing Protein". http://www.iflscience.com/health-and-medicine/scientists-find-way-deactivate-most-common-cancer-causing-protein.
- Downward J (January 2003). "Targeting RAS signalling pathways in cancer therapy". Nat. Rev. Cancer 3 (1): 11–22. doi:10.1038/nrc969. PMID 12509763. https://dx.doi.org/10.1038%2Fnrc969
- Chavda, Burzin; Arnott, John A.; Planey, Sonia Lobo (September 2014). "Targeting protein palmitoylation: selective inhibitors and implications in disease". Expert Opinion on Drug Discovery 9 (9): 1005–1019. doi:10.1517/17460441.2014.933802. ISSN 1746-045X. PMID 24967607. https://dx.doi.org/10.1517%2F17460441.2014.933802

